
Abstract
Duchene muscular dystrophy (DMD) is a progressive muscle wasting disease, caused by mutations in the dystrophin (DMD) on the X chromosome. One-third of patients are estimated to have de novo mutations. To provide in-depth genetic counseling, the comprehensive identification of mutations is mandatory. However, many DMD patients did not undergo genetic diagnosis because detailed genetic diagnosis was not available or their mutational types were difficult to identify. Here we report the genetic testing of a sporadic DMD boy, who died >20 years previously. Dried umbilical cord preserved for 38 years was the only available source of genomic DNA. Although the genomic DNA was severely degraded, multiplex ligation-dependent probe amplification analysis was performed but no gross mutations found. Sanger sequencing was attempted but not conclusive. Next-generation sequencing (NGS) was performed by controlling the tagmentation during library preparation. A nonsense mutation in DMD (p.Arg2095*) was clearly identified in the proband. Consequently, the identical mutation was detected as an 11% mosaic mutation from his healthy mother. Finally, the proband’s sister was diagnosed as a non-carrier of the mutation. Thus using NGS we have identified a pathogenic DMD mutation from degraded DNA and low-level somatic mosaicism, which would have been overlooked using Sanger sequencing.
Similar content being viewed by others

Introduction
Duchene muscular dystrophy (DMD; OMIM 310200) is an X-linked recessive disorder that affects approximately 1 in 3500 live male newborns. DMD patients present severe muscle weakness and they lose their ability to walk before the age of 12 years, coupled with deterioration in respiratory and cardiac functions.1 DMD is caused by mutations in the dystrophin gene (DMD), located at Xp21.2, which is 2.4 Mb in size, and consists of 79 exons that form a 14-kb mRNA transcript.2 Introduction of the quantitative PCR-based technique of multiplex ligation-dependent probe amplification (MLPA), which can amplify all 79 exons of the DMD and is commercially available, has significantly improved mutation detection of exon deletions or duplications.3, 4, 5, 6, 7 Using MLPA, approximately 70% of DMD patients have been shown to have partial deletions or duplications.8 However, Sanger sequencing identification of small mutations within DMD, including small insertions or deletions within an exon (~7%), single-nucleotide point mutations (~20%) and splice site intronic mutations (<1%), requires great effort with associated time and cost issues owing to the size and number of exons of DMD.7, 8 Recently, targeted next-generation sequencing (NGS) approaches have identified previously unknown causative genes for Mendelian disorders.9 Disease panels for NGS are now widely used in the clinical field.10 It is likely that genetic analysis of small DMD mutations will soon be replaced by NGS sequencing. Moreover, because of progress in NGS, more and more diseases with mosaic mutations have been identified, including AKT1 in Proteus syndrome,11 IDH1 in Maffucci syndrome12 and GNAQ in Sturge Weber syndrome.13 Although somatic mosaicism is caused by many factors such as DNA mutations, epigenetic factors and chromosomal abnormalities, its influence on the clinical phenotype and transmission to the next generation is not negligible.
Here we report a sporadic case with a deceased, phenotypically diagnosed but genetically undiagnosed DMD patient. Using NGS, we identified a pathogenic nonsense mutation in DMD from dried umbilical cord. Here we report a sporadic case of a deceased, phenotypically diagnosed but genetically undiagnosed DMD patient by using dried umbilical cord for NGS. We also detected low-level somatic mosaic mutation in the proband’s mother.
Materials and Methods
Consultee
A 36-year-old woman, the sister of the proband, visited our genetic counseling unit to have a carrier genetic test for DMD. The consultee wanted to know whether she had inherited the DMD mutation from her mother as she knew that her elder brother had DMD (Figure 1a). Because mutation analysis was not performed on the proband, examining the mutation in her genome was difficult. First, MLPA was performed, but no gross mutations were identified. This suggested that if she was a carrier, there were only small mutations in her genomic DNA. Identifying single-nucleotide changes in females is difficult and indicated the need for mutational analysis in the proband.

Familial pedigree. (a) Familial pedigree. Squares, males; circles, females; open shape, unaffected; filled shape, DMD patient. (b) Haplotype analysis between the proband (using umbilical cord DNA) and his parents. All five markers on chromosome 9 were inherited from both parents.
Proband
The proband was a Japanese boy, born 38 years previously. He was diagnosed with DMD because of clinical manifestations at 5 years of age. The case had no familial background (Figure 1a), and his mother had no symptoms, such as muscle cramps or high serum creatine kinase. None of the maternal relatives presented with muscle weakness. The proband had a healthy younger sister (the consultee). He passed away from cardiomyopathy in 1991, aged 14 years. Genetic diagnosis was not performed.
Genetic analysis of the family
The diseased boy, his sister and their parents were analyzed for genetic mutations. Informed consent was obtained from the proband’s family, and the experimental protocols and ethical approval for the study were obtained from the Review Boards of Kobe University. Peripheral blood samples were obtained from the proband’s sister and parents. DNA was extracted from these blood samples using the QuickGene Kit (FujiFilm, Tokyo, Japan), following the manufacturer’s instructions. The proband’s DNA was extracted from dried umbilical cord using DNeasy (Qiagen, Venlo, The Netherlands). Initially, deletion or amplification mutations were examined by MLPA (SALSA MLPA; MRC-Holland, Amsterdam, The Netherlands). Following this, point mutations in DMD were detected using the Trusight One Sequencing Panel on the MiSeq platform (Illumina, San Diego, CA, USA). This panel can detect mutations in approximately 4321 genes (http://www.illumina.com/products/trusight-one-sequencing-panel.ilmn), including DMD. For dried umbilical cord, the genomic DNA tagmentation step was shortened to 3 min (instead of 10 min), as recommended in the standard protocol. All procedures were performed according to the manufacturer’s instructions. For quantification and validation of genomic library samples, the Qubit 2.0 Fluorometer system (Life Technologies, Carlsbad, CA, USA) and 2100 Bioanalyzer Instrument (Agilent Technologies, Palo Alto, CA USA) were used. Sequenced data was analyzed using the Variant Studio Software (Illumina), with Homo sapiens data (hg19, build 37.2) for the workflow. The reference genome provides variant annotations and fixes chromosome size in the BAM file output. Each single variant reported in the VCF output file was examined for coverage and Qscore and visualized via the Integrative Genome Viewer (IGV). For variant calling and reporting, the Variant Studio Software was used. Mutations identified as pathological were confirmed by Sanger sequencing following a standard protocol (BigDye Terminator v3.1 Cycle Sequencing Kit, Applied Biosystems by Life Technologies). Sequencing primers for point mutations in the proband were: DMD_exon9_F, 5′-ttactcagcaggcagtctgg-3′ and 5′-tggcacaaatgtcaaaatga-3′. Confirmation of umbilical cord identity was performed using microsatellite markers (D9S306, D9S2105, D9S2170, D9S2171 and D9S21072) on chromosome 9 from the parents’ genome, as reported previously.14
Deep sequencing of genomic DNA from the mothers of sporadic DMD cases
For six sporadic DMD cases with de novo point mutations, peripheral blood samples were also obtained from the patients and their mothers. For deep sequencing of maternal somatic mosaic mutations, 500-bp PCR products harboring suspected mutation sites were purified by gel extraction using the QIAquick Gel Extraction Kit (Qiagen). Each mutation was then analyzed using the Truseq LT PCR-Free Kit (Illumina). All mutations tested in the mothers of sporadic cases and PCR primers are listed in Table 1. In Table 1, depth and frequency of the mutant to consensus allele obtained from ultra-deep sequencing are shown. All procedures and data analysis were performed according to the manufacturer’s instructions.
Results
Genetic test for DMD in the proband and his sister
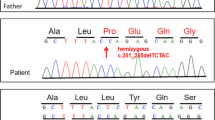
We were unable to obtain genomic DNA from the proband as he had been dead for 23 years. However, the parents offered dried umbilical cord from the deceased patient, as such preservation is a cultural norm for parents in Japan. As the sample had been preserved at room temperature for >38 years, we carefully extracted genomic DNA from the dried umbilical cord. The extracted DNA was degraded to small fragments, ranging from 200 to 3000 bp in size (Figure 2a). Sample identity of the umbilical cord was confirmed using five microsatellite markers on chromosome 9 (Figure 1b). All five markers were found to be inherited from both parents. Next we used MLPA to examine DMD in the proband but detected no exonic deletions or duplications (data not shown). We then attempted to examine all 79 exons by Sanger sequencing but failed to obtain PCR products, especially of longer base lengths (data not shown), which was attributed to genomic DNA degradation. Subsequently, NGS was chosen to examine DMD in the proband. We usually enzymatically digest genomic DNA to obtain fragmented DNA ranging in size from 200 to 1000 bp. However, the degraded genomic DNA had already been naturally fragmented (Figure 2a). Therefore, by shortening the enzyme step for the degraded genomic sample, we were able to obtain a DNA sample of sufficient quantity and quality from the proband for NGS analysis. The proband’s DNA sample had a similar smear pattern (DNA sizes between 200 and 1000 bp) as a control (Figure 2b). NGS identified many nucleotide changes within DMD, but c.6292>T was considered to be the strongest candidate (Figure 3a). Indeed, this change has already been reported as a pathogenic mutation for DMD in the Leiden database (http://www.dmd.nl/). Consequently, we concluded that a hemizygous point mutation (c.6292C>T, p.Arg2095*) in DMD caused DMD in the proband. This nucleotide change was confirmed by Sanger sequencing (Figure 3b). Moreover, also using NGS, we found the same heterozygous somatic mosaic mutation in the mother, at an 11% mutation rate within the total depth (29 out of the 265 depths) detected by the Trusight One Sequencing Panel (Figure 3a). Sanger sequencing showed slight elevation of the mutation allele, although this may not represent a mosaic mutation and instead be considered a nonspecific wave (Figure 3b). Using these results, the patient’s sister was tested for carrier analysis and found to be free of the mutation (Figure 3b).

Genomic DNA and library preparation from dried umbilical cord. (a) Degraded genomic DNA from dried umbilical cord. Electrophoresed genomic DNA. Note smeared degraded DNA of 200– 3000 bp. Lane 1, DNA size marker; Lane 2, genomic DNA from umbilical cord; Lane 3, genomic DNA from peripheral blood. (b) Electrophoresed tagmented DNA. Both degraded genomic DNA and fresh genomic DNA are enzymatically digested to the expected fragment sizes. Smeared bands are located within 200–1000 bp. Lane 1, DNA size marker; Lane 2, tagmented DNA from genomic DNA from peripheral blood; Lane 3, tagmented genomic DNA from dried umbilical cord. bp, base pairs.

Mutation analysis of the proband and his mother. (a) IGV picture of the proband (upper lane) and mother (lower lane). Pathogenic DMD mutation (c.6292C>T, p.Arg2095*) in the proband. Eleven percent mutation rates (29 out of the 265 depths) of the same somatic mosaic mutation was also detected in blood-derived maternal genomic DNA. (b) Sanger sequencing of the pathogenic DMD mutation. Note the slightly elevated mutant allele wave (T) (closed arrow) in the mother.
DMD ultra-deep sequencing detects mosaicism in the mothers of six sporadic DMD cases
Our maternal results indicate that non-carrier mothers of DMD cases may actually be mosaic carriers. Therefore, we additionally tested the mothers of sporadic DMD cases carrying point mutations. Genomic DNA from six mothers (including the present case) was analyzed by NGS using targeted ultra-deep DMD sequencing of the DMD mutation site (500 bp around the mutation site; Table 1). In the present case, we detected 11% of mosaic mutations (1017/9437 depth) in DMD, which is exactly the same mosaic rate as our previous result (29/265 depth, Figure 3a) obtained with the Trusight One disease panel. Thus this suggests that the mosaic rate obtained by the Trusight One disease panel is not coincidentally biased. No mutations were detected in the other five mothers of sporadic cases (Table 1).
Discusssion
Although mutational analysis of point or small DMD mutations has already been vigorously performed, there are many undiagnosed patients with DMD but negative MLPA results. Recently, more and more mutational analyses are reported using NGS. Moreover, NGS disease panels such as Trusight One enable identification of pathogenic mutations when the genetic analysis is not performed in clinically diagnosed or even undiagnosed patients by relatively simple methods. Especially in large genes such as DMD, myosin and titin, reports on pathogenic mutation analysis using NGS have increased.15 Regarding the clinical use of NGS analysis, we informed the patient on certain issues. First, as there are thousands of DMD variants that are not likely pathogenic or with unknown significance, we discussed the limitation of accuracy and validation of the test result. For this study, we were able to confirm a validated pathogenic variant in the proband to the patient’s family because the variant p.Arg2095* is a definite pathogenic mutation. Second, because of the limitation and nature of this study, we did not attempt to detect other gene variants not related to muscular dystrophy. Third, if we find incidental findings, we will discuss the ethics and benefit for the patient with an appropriate board of Kobe University.
It is estimated that more unaffected, female, DMD carriers will consult genetic counseling units for genetic carrier testing because of their family plans. Prenatal genetic testing for DMD is also performed for children of DMD patients or carrier mothers. The a priori carrier risk for mothers of sporadic DMD patients is 66%.16 The presence of unaffected sons and brothers, in combination with low maternal blood creatine kinase levels, reduces this risk. Carrier detection and first trimester prenatal diagnosis has attained >99% reliability in every case.16 Therefore, accurate diagnosis for the proband and probands’ mothers is crucial.
In this report, we used NGS in a sporadic DMD case to genetically diagnose a proband with DMD. Using this information, we could perform carrier genetic testing for a potential female carrier in a family. As a tissue sample (for example, peripheral blood or skeletal muscle) from the proband was unavailable, we extracted genomic DNA from dried umbilical cord, which the majority of Japanese families keep as a memento of their child’s birth. Although the DNA quality was not good enough to amplify long PCR products for Sanger sequencing, shorter (around 300 bp in size) DNA fragments were at least suitable for preparing NGS libraries for subsequent sequencing. Indeed, we failed to amplify PCR products of some exons within DMD. Given that the degraded DNA quality is below the 50% range required for customized PCR sequencing primers, re-arranging the primers for DMD sequencing may cost at least $500. Sequencing 79 exons by Sanger sequencing costs at least $1500. Moreover, validating and re-examining PCRs for failed samples would also be time-consuming. In contrast, a commercially available genetic disease gene panel for NGS analysis encompasses all DMD exonic regions and costs no more than $1000 to detect pathogenic variants in only 4 days of lab experiments and with detailed data analysis using updated software. Therefore, it is more straightforward, faster and cost-effective to use NGS tools instead of conventional methods. To our knowledge, there have been no reports that have tested dried umbilical cord for identifying pathogenic mutations by NGS. As genetic testing has been widely accepted worldwide, the need for carrier genetic testing of small DMD mutations will likely increase. This practice may be beneficial in Japan for genetic diagnosis of deceased patients.
Deep sequencing by NGS has recently detected very-low-grade somatic mosaicism in unaffected mothers of siblings with nemaline myopathy.17 Thus NGS enables deep resequencing of previously undetected low-grade mosaic mutations by Sanger sequencing. The background baseline signal noise of bases in Sanger sequencing can sometimes be confusing or misleading for identifying true variants. Especially in cases with low-grade somatic mosaicism, Sanger methods have limitations for accurately diagnosing mosaic rates. If the mosaic rate is low and/or the background baseline signal is high, mosaic mutations are likely to have been overlooked by Sanger methods. Germline mosaicism was previously suggested if a recurrent DMD patient is born to a non-carrier mother. Therefore, even if a de novo mutation is identified in the proband, prenatal genetic testing for the next child has sometimes been indicated for the pregnant mother. Germline DMD mutations can be problematic in determining recurrent from sporadic DMD cases in families.18 Somatic mosaicism may not have the same rate as germline mosaicism, but NGS may be a useful tool for diagnosing maternal low-grade mosaic mutations, especially in sporadic DMD cases. If low-grade mosaicism were found in such cases, sisters of the proband could also be carriers for the mutations. As DMD is the largest gene, it is not surprising to find somatic mosaic variants over the course of a lifetime. Although we did not detect somatic pathogenic mutations in an additional five mothers, genetic testing by NGS will allow a more correct diagnosis in terms of recurrent risk evaluation in genetic counseling for genetic diseases.
References
Bushby, K., Finkel, R., Birnkrant, D. J., Case, L. E., Clemens, P. R., Cripe, L. et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: implementation of multidisciplinary care. Lancet Neurol. 9, 177–189 (2010).
Hoffman, E. P., Brown, R. H. Jr & Kunkel, L. M. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51, 919–928 (1987).
White, S., Kalf, M., Liu, Q., Villerius, M., Engelsma, D., Kriek, M. et al. Comprehensive detection of genomic duplications and deletions in the DMD gene, by use of multiplex amplifiable probe hybridization. Am. J. Hum. Genet. 71, 365–374 (2002).
Gatta, V., Scarciolla, O., Gaspari, A. R., Palka, C., De Angelis, M. V., Di Muzio, A. et al. Identification of deletions and duplications of the DMD gene in affected males and carrier females by multiple ligation probe amplification (MLPA). Hum. Genet. 117, 92–98 (2005).
Lalic, T., Vossen, R. H., Coffa, J., Schouten, J. P., Guc-Scekic, M., Radivojevic, D. et al. Deletion and duplication screening in the DMD gene using MLPA. Eur. J. Hum. Genet. 13, 1231–1234 (2005).
Janssen, B., Hartmann, C., Scholz, V., Jauch, A. & Zschocke, J. MLPA analysis for the detection of deletions, duplications and complex rearrangements in the dystrophin gene: potential and pitfalls. Neurogenetics 6, 29–35 (2005).
Aartsma-Rus, A., Van Deutekom, J. C., Fokkema, I. F., Van Ommen, G. J. & Den Dunnen, J. T. Entries in the Leiden Duchenne muscular dystrophy mutation database: an overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve 34, 135–144 (2006).
Takeshima, Y., Yagi, M., Okizuka, Y., Awano, H., Zhang, Z., Yamauchi, Y. et al. Mutation spectrum of the dystrophin gene in 442 Duchenne/Becker muscular dystrophy cases from one Japanese referral center. J. Hum. Genet. 55, 379–388 (2010).
Ng, S. B., Buckingham, K. J., Lee, C., Bigham, A. W., Tabor, H. K., Dent, K. M. et al. Exome sequencing identifies the cause of a mendelian disorder. Nat. Genet. 42, 30–35 (2010).
Wei, X., Dai, Y., Yu, P., Qu, N., Lan, Z., Hong, X. et al. Targeted next-generation sequencing as a comprehensive test for patients with and female carriers of DMD/BMD: a multi-population diagnostic study. Eur. J. Hum. Genet. 22, 110–118 (2014).
Lindhurst, M. J., Sapp, J. C., Teer, J. K., Johnston, J. J., Finn, E. M., Peters, K. et al. A mosaic activating mutation in AKT1 associated with the Proteus syndrome. N. Engl. J. Med. 365, 611–619 (2011).
Amary, M. F., Damato, S., Halai, D., Eskandarpour, M., Berisha, F., Bonar, F. et al. Ollier disease and Maffucci syndrome are caused by somatic mosaic mutations of IDH1 and IDH2. Nat. Genet. 43, 1262–1265 (2011).
Shirley, M. D., Tang, H., Gallione, C. J., Baugher, J. D., Frelin, L. P., Cohen, B. et al. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N. Engl. J. Med. 368, 1971–1979 (2013).
Kondo-Iida, E., Kobayashi, K., Watanabe, M., Sasaki, J., Kumagai, T., Koide, H. et al. Novel mutations and genotype-phenotype relationships in 107 families with Fukuyama-type congenital muscular dystrophy (FCMD). Hum. Mol. Genet. 8, 2303–2309 (1999).
Chauveau, C., Rowell, J. & Ferreiro, A. A rising titan: TTN review and mutation update. Hum. Mutat. 35, 1046–1059 (2014).
Bakker, E., Veenema, H., Den Dunnen, J. T., van Broeckhoven, C., Grootscholten, P. M., Bonten, E. J. et al. Germinal mosaicism increases the recurrence risk for 'new' Duchenne muscular dystrophy mutations. J. Med. Genet. 26, 553–559 (1989).
Miyatake, S., Koshimizu, E., Hayashi, Y. K., Miya, K., Shiina, M., Nakashima, M. et al. Deep sequencing detects very-low-grade somatic mosaicism in the unaffected mother of siblings with nemaline myopathy. Neuromuscul. Disord. 24, 642–647 (2014).
Helderman-van den Enden, A. T., de Jong, R., den Dunnen, J. T., Houwing-Duistermaat, J. J., Kneppers, A. L., Ginjaar, H. B. et al. Recurrence risk due to germ line mosaicism: Duchenne and Becker muscular dystrophy. Clin. Genet. 75, 465–472 (2009).
Acknowledgements
We greatly appreciate the patients and their family’s cooperation for this study. We thank Mrs Tomomi Emoto for technical assistance. This work was supported by a donation from Kobe city to the Department of General Pediatrics, Kobe University Graduate School of Medicine (K550003302).
Author contributions
MT-I took care of the whole family for genetic counseling, performed the experiments and wrote the manuscript. HN and YT took care of the proband and wrote the manuscript. TL, MY, HA, MN, AU and KN performed genetic analysis of the DMD patients and wrote the manuscript. IM, MM, TT, HK and KI were involved in planning the whole project and wrote the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Taniguchi-Ikeda, M., Takeshima, Y., Lee, T. et al. Next-generation sequencing discloses a nonsense mutation in the dystrophin gene from long preserved dried umbilical cord and low-level somatic mosaicism in the proband mother. J Hum Genet 61, 351–355 (2016). https://doi.org/10.1038/jhg.2015.157
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2015.157
This article is cited by
-
EMQN best practice guidelines for genetic testing in dystrophinopathies
European Journal of Human Genetics (2020)
-
Targeted next-generation sequencing-based molecular diagnosis of congenital hand malformations in Chinese population
Scientific Reports (2018)
-
Cryptic splice activation but not exon skipping is observed in minigene assays of dystrophin c.9361+1G>A mutation identified by NGS
Journal of Human Genetics (2017)
