
Abstract
Agnathia-otocephaly complex is a malformation characterized by absent/hypoplastic mandible and abnormally positioned ears. Mutations in two genes, PRRX1 and OTX2, have been described in a small number of families with this disorder. We performed clinical and genetic testing in an additional family. The proband is a healthy female with a complicated pregnancy history that includes two offspring diagnosed with agnathia-otocephaly during prenatal ultrasound scans. Exome sequencing was performed in fetal DNA from one of these two offspring revealing a heterozygous duplication in OTX2: c.271_273dupCAG, p.(Gln91dup). This change leads to the insertion of a glutamine within the OTX2 homeodomain region, and is predicted to alter this signaling molecule’s ability to interact with DNA. The same variant was also identified in the proband’s clinically unaffected 38-year-old husband and their 9-year-old daughter, who presented with a small mandible, normal ears and velopharyngeal insufficiency due to a short hemi-palate. This unusual presentation of OTX2-related disease suggests that OTX2 might have a role in palatal hypoplasia cases. A previously unreported OTX2 variant associated with extreme intrafamilial variability is described and the utility of exome sequencing as a tool to confirm the diagnosis of agnathia-otocephaly and to inform the reproductive decisions of affected families is highlighted.
Similar content being viewed by others


Main
OTX2 (MIM*600037) encodes a transcription factor with essential functions in embryonic head formation and, at later developmental stages, in eye and brain development.1 After birth, although its expression decreases, it maintains an important role in the retina.2 The OTX2 protein is a member of the OTX group of signaling molecules, a highly conserved protein subclass characterized by a DNA-binding homeodomain region.1
Defects in OTX2 can lead to a wide range of phenotypes. OTX2 haploinsufficiency, resulting from heterozygous loss-of-function mutations, is typically associated with structural eye defects (ranging from bilateral anophthalmia to coloboma).3 Ocular abnormalities are often combined with extraocular features including learning disability and pituitary abnormalities.1, 3 OTX2-related disorders that do not primarily involve the eye have also been reported. These include hemifacial microsomia (that is, facial asymmetry due to maxillary and mandibular hypoplasia and ear malformations), and the agnathia-otocephaly complex (AOC).4, 5, 6, 7
AOC (MIM#202650) is a rare, often sporadic, malformation complex of the first pharyngeal arch that is characterized by agnathia/dysgnathia, microstomia, aglossia/hypoglossia and variable displacement of the ears toward the midline. Prognosis is poor with affected individuals often dying shortly after birth due to respiratory failure secondary to airway obstruction.7 Nevertheless, a few patients have been reported to survive into childhood. AOC has been attributed to both genetic and teratogenic causes.5, 6, 7, 8 Genetic causes include heterozygous or biallelic PRRX1 mutations as well as heterozygous, presumed loss-of-function OTX2 mutations (Table 1).5, 6, 8
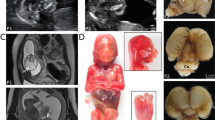
This study expands the phenotypic spectrum of OTX2-related disease and details the clinical and genetic findings in a family segregating a previously unreported three base-pair duplication in OTX2 (Figure 1a). The proband is a healthy female with a complicated obstetric history (II:2): during her first pregnancy, a 23-week ultrasound scan revealed a fetus with a small jaw and low-set ears (III:1). There was polyhydramnios and she went into preterm labor; there was immediate neonatal death and the diagnosis of AOC was confirmed on autopsy. A subsequent pregnancy (III:5) was electively terminated after a 17-week scan revealed polyhydramnios and facial malformations suggestive of recurrence of AOC. DNA was extracted from fetal (III:5) tissue and analyzed using exome sequencing (Supplementary Information). A heterozygous c.271_273dupCAG, p.(Gln91dup) change in OTX2 was identified. This change was also found in the proband’s clinically unaffected 38-year-old husband (II:1) and their 9-year-old daughter (III:2; Figure 2). The latter presented with respiratory problems after birth and was noted to have a small mandible. Intra-oral examination revealed an unusually short left hemi-palate. She failed to thrive because of feeding difficulties and was fed by nasogastric tube for the first year of life. No other malformations were noted and the diagnosis of AOC was excluded. Although she started to speak at the normal time, her speech was severely hypernasal. A videofluoroscopic swallowing study at age 3 years confirmed a hypoplastic soft palate on the left side; there was only a small amount of palate movement on the right side and no movement on the left side. A Furlow palatoplasty was carried out at the age of 3 years followed by a hemi-pharyngoplasty at the age of 6 years, and these resulted in a marked improvement in her speech quality. At the age of 9 years, there are no concerns about her health and development; she is moderately myopic and has a normal eye examination.

(a) Pedigree of the reported family. Subject III:1 was diagnosed with likely agnathia-otocephaly complex during a prenatal ultrasound scan; there was immediate neonatal death after preterm labor at 30 weeks gestation. Subject III:2 has micrognathia and had surgery to correct her hypoplastic left soft palate. For subject III:5, there was medical termination of pregnancy after prenatal ultrasonography revealed recurrence of agnathia-othocephaly complex. Subject II:1 was found to have the c.271_273dupCAG, p.(Gln91dup) mutation but he is unaffected (no palatal abnormalities and just a mild impression of having a small jaw). The proband (II:2) was also unaffected and did not harbor the OTX2 duplication. (b) Predicted protein structure of the OTX2 homeodomain. Overlay of normal (green) and c.271_273dupCAG, p.(Gln91dup) mutant (yellow) protein is presented. Similar to other transcription factors, the OTX2 homeodomain comprises a 60 amino-acid region (corresponding to amino acids 46–105 (NP_068374.1)) in which three α-helices (shown as cylinders) are connected by short loops. The more C-terminal helix of this extremely conserved structure is the longest and lies roughly perpendicular to the other two. It is known as the recognition helix as it confers susceptibility to DNA binding (schematic in top left hand corner panel). Previous functional studies have emphasized the critical role of three residues of the recognition helix in setting sequence-specific protein–DNA contacts.9 These are valine 92, lysine 95 and asparagine 96 in OTX2 (NP_068374.1; highlighted). The mutation identified in the family adds a glutamine at position 91 and distorts the recognition helix close to these three, critical for DNA recognition, residues (yellow protrusion from helix). It is also possible that an insertion like this will not disturb the helical structure but it will shift all subsequent residues by one position (i.e., one third of the way around the helix). The latter could lead to loss of binding; the side chains will rotate so that the residues pointing out toward the DNA will be pointing in the wrong direction for making interactions. The structural model was generated by using the SWISS-MODEL protein homology modeling server (Swiss Institute of Bioinformatics and the Biozentrum University of Basel, Switzerland). The solution structure of the OTX2 homeobox domain with Protein Data Bank ID 2dms.1 was utilized. PyMOL (PyMOL Molecular Graphics System, Version 1.3r1, Schrödinger, LLC) was used to view the three-dimensional molecular structures. A full color version of this figure is available at the Journal of Human Genetics journal online.

Clinical photographs of two related individuals harboring a heterozygous c.271_273dupCAG change in OTX2. Subject II:1 (a) is clinically unaffected while his daughter, subject III:2, (b) presented with an unusually short left hemi-palate and severe micrognathia at birth. A full color version of this figure is available at the Journal of Human Genetics journal online.
The c.271_273dupCAG, p.(Gln91dup) change affects the 3′ end of OTX2 exon 4 (found in both major OTX2 isoforms (NP_068374.1 and NP_758840.1)). This duplication does not disrupt the sequence of the splice donor site and it was not found to have a significant effect on splicing by in silico analysis (Alamut v2.2.1, Interactive Biosoftware, Rouen, France). However, it would lead to the insertion of a glutamine residue, within the highly conserved, from fly to human homeodomain region.9 We have evaluated the physiological significance of this change using homology modeling and the results are presented in Figure 1b.
Over 50 disease-associated variants have been reported in OTX2 (HGMD, Cardiff, UK, accessed 31 October 2014). These are sparsely distributed throughout the gene and typically result in a protein product with reduced or no function.1, 4 All cases with OTX2-related disease reported to date were heterozygotes with 40% known to be de novo mutational events. Variable expressivity is common.3 We report four related cases harboring the same OTX2 variant and presenting with different phenotypes: one has a normal examination and just a mild impression of having a small jaw (II:1; Figure 2a), one has an abnormally small jaw (Figure 2b) and a hypoplastic left soft palate (III:2) and two were severely affected with AOC (III:5 and III:1; no molecular confirmation in the latter). Significant intrafamilial variability has been previously reported in two families with OTX2-related AOC,5, 6 but neither a clinically unaffected individual nor a subject with a phenotype similar to that of subject III:2 have been previously described (Table 1). Notably, incomplete penetrance has been reported in families segregating OTX2-associated structural eye defects.3
AOC is genetically heterogeneous and mutations in two genes, PRRX1 (recessive or dominant) and OTX2 (dominant), explain only a small subset of cases.5, 6, 7, 8 The condition is typically sporadic and often suspected during second trimester ultrasound scans.7 We have demonstrated the utility of exome sequencing as a means of elucidating the molecular pathology in fetuses with AOC. Knowledge of the genetic diagnosis allows more accurate estimation of the recurrence risk and can guide the reproductive decisions of affected families.
An unusual presentation of OTX2-related disease consisting of migrognathia and unilateral soft palate hypoplasia is described. Ongoing work to dissect the major roles and key interactions of this important transcription factor may allow identification of modifier genes and shed light on the mechanisms that underlie variable expressivity.
References
Beby, F. . & Lamonerie, T. The homeobox gene Otx2 in development and disease. Exp. Eye Res. 111, 9–16 (2013).
Roger, J. E., Hiriyanna, A., Gotoh, N., Hao, H., Cheng, D. F. . & Ratnapriya, R. et al. OTX2 loss causes rod differentiation defect in CRX-associated congenital blindness. J. Clin. Invest. 124, 631–643 (2014).
Williamson, K. A. . & FitzPatrick, D. R. The genetic architecture of microphthalmia, anophthalmia and coloboma. Eur. J. Med. Genet. 57, 369–380 (2014).
Zielinski, D., Markus, B., Sheikh, M., Gymrek, M., Chu, C. . & Zaks, M. et al. OTX2 duplication is implicated in hemifacial microsomia. PLoS ONE 9, e96788 (2014).
Chassaing, N., Sorrentino, S., Davis, E. E., Martin-Coignard, D., Iacovelli, A. . & Paznekas, W. et al. OTX2 mutations contribute to the otocephaly-dysgnathia complex. J. Med. Genet. 49, 373–379 (2012).
Patat, O., van Ravenswaaij-Arts, C. M., Tantau, J., Corsten-Janssen, N., van Tintelen, J. P. . & Dijkhuizen, T. et al. Otocephaly-dysgnathia complex: description of four cases and confirmation of the role of OTX2. Mol. Syndromol. 4, 302–305 (2013).
Gekas, J., Li, B. . & Kamnasaran, D. Current perspectives on the etiology of agnathia-otocephaly. Eur. J. Med. Genet. 53, 358–366 (2010).
Donnelly, M., Todd, E., Wheeler, M., Winn, V. D. . & Kamnasaran, D. Prenatal diagnosis and identification of heterozygous frameshift mutation in PRRX1 in an infant with agnathia-otocephaly. Prenat. Diagn. 32, 903–905 (2012).
Chatelain, G., Fossat, N., Brun, G. . & Lamonerie, T. Molecular dissection reveals decreased activity and not dominant negative effect in human OTX2 mutants. J. Mol. Med. (Berl) 84, 604–615 (2006).
Acknowledgements
We would like to thank the family for their participation and assistance. This research was supported by the Manchester Biomedical Research Centre and NIHR Greater Manchester: Clinical Research Network.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Sergouniotis, P., Urquhart, J., Williams, S. et al. Agnathia-otocephaly complex and asymmetric velopharyngeal insufficiency due to an in-frame duplication in OTX2. J Hum Genet 60, 199–202 (2015). https://doi.org/10.1038/jhg.2014.122
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2014.122
This article is cited by
-
Re-focusing on Agnathia-Otocephaly complex
Clinical Oral Investigations (2021)
-
A de novo variant in OTX2 in a lamb with otocephaly
Acta Veterinaria Scandinavica (2020)
-
Genetics of anophthalmia and microphthalmia. Part 2: Syndromes associated with anophthalmia–microphthalmia
Human Genetics (2019)
-
Prenatal Diagnostic Exome Sequencing: a Review
Current Genetic Medicine Reports (2017)
