
Abstract
Five new secondary metabolites derived from pentanol, namely catathelasmols A–E (1–5), were isolated from the fruiting bodies of the basidiomycete Catathelasma imperiale. Their structures were elucidated on the basis of spectroscopic analysis, and the absolute configurations were determined by computational chemistry. Compounds 3, 4 and 5 showed inhibitory activities against two isozymes of 11-hydroxysteroid dehydrogenases (11-HSD1 and 11-HSD2), with IC50 values of 28.7–62.3 g ml−1 (human 11-HSD1), 30.4–149.2 g ml−1 (mouse 11-HSD1), 5.1–177 g ml−1 (human 11-HSD2) and 32.3–129.1 g ml−1 (mouse 11-HSD2), which catalyze the interconversion of cortisol and cortisone.
Similar content being viewed by others

Introduction
Catathelasma imperiale (Fr.) Sing. (Tricholomataceae) is a conifer-loving basidiomycete defined by its large size, white spore print, sticky brownish cap, mealy odor and double ring. This mushroom is mainly distributed in the southwest of China. Only a few ergosterols have been reported from the chemical investigation of this fungus.1 As a part of our efforts to discover the structurally diverse and biologically active secondary metabolites from higher fungi,2, 3, 4, 5 the investigation of the fruiting bodies of C. imperiale has led to the isolation of five new compounds, catathelasmols A–E (1–5). Herein, details of the isolation and structural elucidation of 1–5 are described, including assignment of the absolute configurations by computational chemistry. Among them, compounds 3–5 showed inhibitory activities against two isozymes of 11β-hydroxysteroid dehydrogenases (11β-HSD1 and 11β-HSD2), which catalyze the interconversion of cortisol and cortisone. Although these secondary metabolites are structurally simple, there are very few reports on these types of natural products.
Materials and methods
General
Optical rotations were obtained on a Horiba SEPA-300 polarimeter (Horiba, Tokyo, Japan). IR spectra were taken on a Bruker Tensor 27 FT-IR spectrometer (Bruker GmbH, Ettlingen, Germany) with KBr pellets. NMR spectra were recorded with a Bruker DRX-500 instrument (Bruker GmbH) in CDCl3 (δH=7.26 p.p.m., δC=77.00 p.p.m.) at room temperature. EI-MS, electrospray ionization mass spectrum (ESI-MS) and high resolution electrospray ionization mass spectrum (HR-ESI-MS) were measured on Finnigan-MAT 90 (Finnigan, Somerset, NJ, USA) and API QSTAR Pulsar i (MDS Sciex, Concord, ON, Canada) mass spectrometers, respectively. Silica gel (200–300 mesh; Qingdao Marine Chemical Inc., Qingdao, China) and Sephadex LH-20 (Amersham Biosciences, Uppsala, Sweden) were used for column chromatography. Fractions were monitored by TLC and spots were visualized by heating silica gel plates sprayed with vanillin-H2SO4 in ethanol.
Fungus material
The fresh fruiting bodies of C. imperiale were purchased at a market in Nanhua County of Yunnan Province, China, in August 2005 and were identified by Professor Mu Zang. The voucher specimen (HFG 05112) was deposited in the herbarium of Kunming Institute of Botany, Chinese Academy of Sciences.
Extraction and isolation
The dry fruiting bodies of C. imperiale (700 g) were extracted thrice with EtOAc (total 9 l) at room temperature for 3 days each time. The extract was filtered and concentrated under reduced pressure to give a residue (18.8 g), which was subjected to silica gel column chromatography eluted with CHCl3/MeOH (from 100:0 to 0:100). The fraction (6.6 g) eluted with pure CHCl3 was subjected to further silica gel column chromatography using a gradient of petroleum ether:acetone (150:1; 50:1; 20:1) followed by pure MeOH to give subfractions A–D. Subfraction A (300 mg) (petroleum ether:acetone 150:1, v/v) was further isolated over a silica gel column eluted with petroleum ether:acetone (150:1) to give a residue (160 mg) mainly containing 1 and 2, which was repeatedly subjected to silica gel column chromatography to yield 1 (40.0 mg) and 2 (1.7 mg). Subfraction B (180 mg) (petroleum ether:acetone 50:1, v/v), containing mainly 3 and 4, was further isolated on a silica gel column eluted with CHCl3 to yield 3 (14.6 mg) and 4 (74.1 mg). Subfraction D (121 mg) eluted with MeOH was purified by Sephadex LH-20 (CHCl3/MeOH 1:1, v/v) and silica gel column chromatography using CHCl3/MeOH (150:1, v/v) as the eluent, to yield 5 (61.4 mg).
Computational methods
The stable geometries of 1 with low energy were investigated using HyperChem 7.0 (HyperCube, Gainesville, FL, USA). These low-energy conformations were then optimized at the B3LYP/6-31G (d) level again. The B3LYP/6-31G (d)-optimized structures were then used for optical rotation calculations at the B3LYP/aug-cc-pVDZ//6-31+G (d). The calculated optical rotation for (R) configuration was +14.5°. This value is very close to the experimental magnitude of +10.6°. Thus, the absolute configuration of (+)-1 was assigned as (R). Compound 5 has a linear structure and it is difficult to use the above method to compute its optical rotation to determine the absolute configuration. Our recent matrix method was used in the study. The value of the determinant (det(D)) for (R)-5 was −2.50. According to the principle of matrix prediction, the det(D) and k0 value were needed to use both. As the optical rotation was −10.4 (c 0.40, CHCl3), the calculated k0 value for this chiral secondary alcohol was 4.2, which is located in the window of coefficients of chiral secondary alcohols in chloroform. Thus, compound 5 was assigned (R) configuration.
Physicochemical properties
Catathelasmol A (1): amorphous powder; [α]D26 +10.6 (c 0.30, CHCl3); IR (KBr): 3450, 1175, 1115, 1058, 1033 cm–1; 1H- and 13C-NMR: see Table 1; ESI-MS (pos.): 101 [M–H2O+H]+, 83 [M–2H2O+H]+; HR-ESI-MS (pos.): 101.0600 ([M–H2O+H]+, calcd. 101.0602).
Catathelasmol B (2): colorless oil; [α]D26 +8.9 (c 0.30, CHCl3); IR (KBr): 3441, 1191, 1167, 1104, 1062, 1034 cm–1; 1H- and 13C-NMR: see Table 1; ESI-MS (pos.): 218 [M]+, 201 [M–H2O+H]+, 183 [M–2H2O+H]+; HR-ESI-MS (pos.): 201.1125 ([M–H2O+H]+, calcd. 201.1126).
Catathelasmol C (3): colorless oil; IR (KBr): 1736, 1661, 1371, 1236, 1191 cm–1. 1H- and 13C-NMR: see Table 2; ESI-MS (pos.): 225 [M+Na]+; HR-ESI-MS (pos.): 225.0739 ([M+Na]+, calcd. 225.0738).
Catathelasmol D (4): colorless oil; IR (KBr): 3441, 2966, 1738, 1369, 1248, 1045 cm−1; 1H- and 13C-NMR: see Table 2; EI-MS: 160 (M+, 2), 129 (42), 112 (17), 100 (14), 87 (100); HR-FAB-MS (pos.): 161.0838 ([M+H]+, calcd. 161.0814).
Catathelasmol E (5): colorless oil; [α]D26 –10.4 (c 0.40, CHCl3); IR (KBr): 3458, 1740, 1450, 1247 cm–1. 1H- and 13C-NMR: see Table 2; ESI-MS (pos.): 227 [M+Na]+; HR-ESI-MS (pos.): 227.0889 ([M+Na]+, calcd. 227.0895).
Biological testing
The inhibitory activities of the compounds on human or mouse 11β-HSD1 and 11β-HSD2 enzymatic activities were determined by the scintillation proximity assay using microsomes containing 11β-HSD1 or 11β-HSD2, according to our earlier studies.6 Briefly, the full-length cDNAs of human or murine 11β-HSD1 and 11β-HSD2 were isolated from the cDNA libraries provided by the NIH Mammalian Gene Collection and cloned into pcDNA3 expression vector. HEK-293 cells were transfected with the pcDNA3-derived expression plasmid and selected by cultivation in the presence of 700 μg ml−1 of G418. The microsomal fraction overexpressing 11β-HSD1 or 11β-HSD2 was prepared from the HEK-293 cells stably transfected with either 11β-HSD1 or 11β-HSD2 and was used as the enzyme source for scintillation proximity assay. Microsomes containing human or mouse 11β-HSD1 were incubated with NADPH and [3H]cortisone. Then the product, [3H]cortisol, was specifically captured by a monoclonal antibody coupled to protein A-coated scintillation proximity assay beads. The 11β-HSD2 screening was performed by incubating 11β-HSD2 microsomes with [3H]cortisol and NAD+ and monitoring substrate disappearance. IC50 values were calculated by using Prism Version 4 (GraphPad Software, San Diego, CA, USA).
Results and discussion
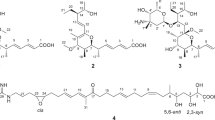
Catathelasmol A (1), obtained as an amorphous powder, has a molecular formula of C5H10O3 based on the positive-ion HR-ESI-MS, showing a quasi-molecular ion peak at m/z 101. 0600 (calcd. for [C5H10O3–H2O+H]+, 101.0602) and requiring only one degree of unsaturation. The IR spectrum showed the presence of one or more hydroxyl groups (3450 cm–1). The 13C-NMR spectrum (Table 1) exhibited five signals: one quaternary carbon bearing two oxygens at δ 102.7, two oxymethylenes at δ 68.0 (t), 65.2 (t) and two up-field methylenes at δ 33.8 (t), 23.5 (t). It was obvious that only one degree of unsaturation was attributed to a ring. The 1H-NMR spectrum (Table 1) showed eight protons: two oxygenated methylenes at δ 3.45 (1H, d, J=11.5 Hz), 4.14 (1H, d, J=11.5 Hz), 3.97 (2H, t, J=6.9 Hz), and up-field resonances at δ 1.61 (1H, m), 1.94 (1H, m), 1.88 (1H, m), 2.05 (1H, m). The above NMR data suggested that 1 possessed a tetrahydrofuran moiety connected with a hydroxyl and a hydroxymethyl group. The heteronuclear multi-bond correlations (HMBC) (Figure 1) from H-6 to C-2 and C-3, and from H-5 to C-2 and C-3 were observed; consequently, the hydroxyl and hydroxymethyl groups were doubtless both emplaced at C-2. The absolute configuration of (+)-1 was assigned as (R) using the B3LYP/aug-cc-pVDZ//6-31+G(d) methods7, 8, 9 based on the comparison of experimental optical rotation (+10.6°) and calculated optical rotation (+14.5°). Therefore, the structure of 1 was determined as (R)-(+)-2-(hydroxymethyl)-tetrahydrofuran-2-ol and named catathelasmol A, as shown in Figure 2.

Key HMBC correlations of compounds 1, 3 and 5.

Structures of compounds 1–5.
Catathelasmol B (2), a colorless oil, was obtained as a minor constituent with a molecular formula of C10H18O5, based on the positive-ion HR-ESI-MS: 201.1125 (calcd. for [C10H18O5–H2O+H]+, 201.1126). In the NMR spectra (Table 1) of 2, signals for the number of protons and carbon that were observed were only half of the number that would correspond to the molecular formula. This indicated that 2 is a symmetrical structure. The NMR data were considerably in accordance with those of 1, but their TLC behavior was discriminable, which suggested that 2 was unambiguously a dimer of 1, and there were just two possible condensed positions: C-2 or C-6 hydroxyl. There was no reaction and no corresponding product obtained in acetylation, which suggested that there was no free primary hydroxyl group in the structure of 2, but that 2 was formed from two molecules of 1 through the intermolecular dehydration at C-6 hydroxyl. Thus, the structure of 2 was proposed as (2R,2′R)-2,2′-oxybis(methylene)bis(tetrahydrofuran-2-ol), as shown in Figure 2.
Catathelasmol C (3) was isolated as a colorless oil possessing the molecular formula C9H14O5, based on the positive-ion HR-ESI-MS: 225.0739 (calcd. for C9H14O5Na, 225.0738). The IR spectrum showed the absorption bands of ester carbonyl (1736 cm1) and keto carbonyl (1661 cm–1) groups. The 13C-NMR spectrum (Table 2) exhibited nine carbon resonances, including one keto carbonyl at δ 202.9 (s), two oxymethylenes at δ 67.9 (t), 66.3 (t), two up-field methylene carbons at δ 35.1 (t), 22.2 (t), as well as characteristic signals at δ 170.2 (s), 20.4 (q); 171.0 (s), 20.9 (q) contributed to two acetoxyls. The 1H-NMR spectrum (Table 2) of 3 showed signals for two oxygenated methylenes at δ 4.65 (2H, s), 4.06 (2H, t, J=6.3 Hz), two up-field methylenes at δ 2.50 (2H, t, J=7.2 Hz), 1.95 (2H, m) together with two acetoxyl methyl singlets at δ 2.16 (3H, s), 2.04 (3H, s). The above NMR character allowed us to conclude that 3 was a diacetylated pentanediol containing a keto group. By analysis of the HMBC spectrum (Figure 1), the position of the ketone was determined at C-2, in which the correlations from H-1 to C-3 and C-a1, from H-5 to C-3 and C-b1 and from H-4 to C-2 were observed. Therefore, the structure of 3 was elucidated as 2-oxopentane-1,5-diyl diacetate named catathelasmol C, which was a new natural product.10
Catathelasmol D (4), also obtained as a colorless oil, has a molecular formula of C7H12O4, based on the EI-MS showing a molecular ion peak at m/z 160, in combination with the 13C-NMR (DEPT) spectrum. The NMR data (Table 2) of 4 were similar to those of 3, but there was only one set of acetoxyl signals: δ 170.9 (s), 20.5 (q). Considering that the signal at δ 4.26 (2H, s, H-1) in 4 evidently shifted up-field (δ=0.39 p.p.m.) compared with that of 3, the acetoxyl group must be connected at C-5 in 4. Thus, the structure of 4 was determined as 5-hydroxy-4-oxopentyl acetate and named catathelasmol D, which was also a new natural product,11 as shown in Figure 2.
Catathelasmol E (5), a colorless oil, was assigned the molecular formula C9H16O5 by the positive-ion HR-ESI-MS: 227.0889 (calcd. for C9H16O5Na, 227.0895). The NMR data of 5 were similar to those of 3, and comparison of 13C-NMR data showed that instead of a ketone, one oxymethine carbon at δ 69.3 (d) newly appeared in 5. The obvious differences in the 1H-NMR spectra were as follows: (a) the oxymethylene singlet in 3 was observed separately from each other at δ 3.95 (1H, dd, J=11.3, 7.2 Hz) and 4.11 (1H, dd, J=11.3, 7.2 Hz), respectively in 5; and (b) an oxygen-bearing methine proton at δ 3.84 (1H, m) was newly detected in 5. The above NMR character indicated that the keto group in 3 was hydrogenated in 5. Initially, the modified Mosher’s method12, 13 was applied to determine the absolute configuration of the hydroxyl at C-2, but failed. Alternatively, we resorted to the computational method to establish the absolute configuration. The absolute configuration of 5 was assigned as (R) using matrix model.14 The det(D) value for this chiral alcohol was −2.50 and the calculated k0 value was 4.2, which is located in the window of coefficients of chiral secondary alcohols in chloroform. Accordingly, the structure of 5 was established as 2-hydroxypentane-1,5-diyl diacetate, shown in Figure 2, and was named catathelasmol E.
It was not until quite recently that such simple pentanol derivatives were found, which suggested that this type of metabolites were distributed in a narrow range, so we believe that they may possess an important chemotaxonomic significance. Besides, the realization of the chemical transformation from 4 to 1 validated the structural correctness (Figure 3).

Chemical transformation from 4 to 1.
Chemical transformation from 4 to 1:15 A solution of 4 (50 mg) in MeOH (5.0 ml) was treated with K2CO3 (97.4 mg) at room temperature. After stirring for 1.5 h, saturated aqueous ammonium chloride was added and extracted with ethyl acetate, and the organic layer was washed with water and brine, dried over sodium sulfate, and evaporated to a residue that was purified by a silica gel column using pure CHCl3 to yield 1 (12 mg). The determined optical rotation was expectedly zero. Thus, the cyclization of 4 to 1 obtained in the experiments must involve the enzyme catalysis.
Glucocorticoid hormones play important roles in many biological and physiological processes, including regulation of energy metabolism, inflammatory, immune and stress responses, and cardiovascular homeostasis. The action of glucocorticoid on target tissue is not dependent inevitably on the circulating levels, but is regulated in a tissue-specific manner by the enzymes of 11β-hydroxysteroid dehydrogenases (11β-HSD1 and 11β-HSD2), which catalyze the interconversion of active 11-hydroxy-glucocorticoids (cortisol in humans and corticosterone in rodents) and their respective inert 11-keto forms (cortisone in humans and 11-dehydrocorticosterone in rodents).16 11β-HSD1 is highly expressed in the liver, gonad, adipose tissue and brain, in which it acts as a reductase regenerating the active glucocorticoids from its inactive forms, thus amplifying local glucocorticoid action.17 11β-HSD2 is predominantly expressed in aldosterone target cells, such as the kidney and colon, in which it catalyzes the inactivation of glucocorticoids, thereby preventing the excessive activation of the mineralocorticoid receptor and sequelae, including sodium retention, hypokalemia and hypertension.
We tested the inhibitory effect of the compounds on both human and mouse 11β-HSD1 and 11β-HSD2. Compound 3 showed inhibitory activities against 11β-HSD1 (human IC50=28.7 μg ml−1; mouse IC50=30.4 μg ml−1) and 11β-HSD2 (human IC50=5.1 μg ml−1; mouse IC50=32.3 μg ml−1). Compound 4 showed inhibitory activities against 11β-HSD1 (human IC50=47.4 μg ml−1; mouse IC50=149.2 μg ml−1) and 11β-HSD2 (human IC50=38.9 μg ml−1; mouse IC50=129.1 μg ml−1). Compound 5 showed inhibitory activities against human 11β-HSD1 (IC50=62.3 μg ml−1) and 11β-HSD2 (IC50=177.0 μg ml−1). Therefore, compounds 3–5 showed inhibitory activities against 11β-HSD1 and 11β-HSD2 and provide the possibility for modulating local cortisone/cortisol availability in vivo.
References
Yang, S. P., Xu, J. & Yue, J. M. Sterols from the fungus Catathelasma imperiale. Chin. J. Chem. 21, 1390–1394 (2003).
Liu, J. K. N-containing compounds of macromycetes. Chem. Rev. 105, 2723–2744 (2005).
Liu, J. K. Natural terphenyls: developments since 1877. Chem. Rev. 106, 2209–2223 (2006).
Zhou, Z. Y. et al. Gallicynoic acids A–I, acetylenic acids from the Basidiomycete Coriolopsis gallica. J. Nat. Prod. 71, 223–226 (2008).
Zhang, L., Wang, F., Dong, Z. J., Steglich, W. & Liu, J. K. A new butenolide-type fungal pigment from the mushroom Pulveroboletus ravenelii. Heterocycles 68, 1455–1458 (2006).
Yang, H. Y., Dou, W., Lou, J., Leng, Y. & Shen, J. H. Discovery of novel inhibitors of 11beta-hydroxysteroid dehydrogenase type 1 by docking and pharmacophore modeling. Bioorg. Med. Chem. Lett. 18, 1340–1345 (2008).
Liu, D. Z. et al. Vibralactone: a lipase inhibitor with an unusual fused beta-lactone produced by cultures of the basidiomycete Boreostereum vibrans. Org. Lett. 8, 5749–5752 (2006).
Amos, R. D. Electric and magnetic properties of CO, HF, HCI, and CH3F. Chem. Phys. Lett. 87, 23–26 (1982).
Polavarapu, P. L. Ab initio molecular optical rotations and absolute configurations. Mol. Phys. 91, 551–554 (1997).
Kuschinsky, G., Lange, G., Scholtissek, C. h. & Turba, F. Reaction mechanism of digitalis constituents. Biochem. Z. 327, 314–330 (1955).
Bonini, C., Chiummiento, L., Funicello, M., Lupattelli, P. & Pullez, M. New functionalized hydroxymethyl ketones from the mild and chemoselective KMnO4 oxidation of chiral terminal olefins. Eur. J. Org. Chem. 80–83 (2006).
Dale, J. A. & Mosher, H. S. Nuclear magnetic resonance enantiomer regents. Configurational correlations via nuclear magnetic resonance chemical shifts of diastereomeric mandelate, O-methylmandelate, and α-methoxy-α-trifluoromethylphenylacetate (MTPA) esters. J. Am. Chem. Soc. 95, 512–519 (1973).
Ohtani, I., Kusumi, T., Kashman, Y. & Kakisawa, H. High-field FT NMR application of Mosher's method. The absolute configurations of marine terpenoids. J. Am. Chem. Soc. 113, 4092–4096 (1991).
Zhu, H. J., Ren, J. & Pittman, C. U. Matrix model to predict specific optical rotations of acyclic chiral molecules. Tetrahedron 63, 2292–2314 (2007).
Yiotakis, A., Magriotisb, P. A. & Vassiliou, S. A simple synthesis of the metabotropic receptor ligand (2S)-α-(hydroxymethyl)-glutamic acid and its Fmoc protected derivatives. Tetrahedron: Asymmetry 18, 873–877 (2007).
Stewart, P. M. & Draper, N. 11β-hydroxysteroid dehydrogenase and the pre-receptor regulation of corticosteroid hormone action. J. Endocrinol. 186, 251–271 (2005).
Seckl, J. R. & Walker, B. R. Minireview: 11beta-hydroxysteroid dehydrogenase type 1-a tissue-specific amplifier of glucocorticoid action. Endocrinology 142, 1371–1376 (2001).
Acknowledgements
This project was supported by National Basic Research Program of China (973 Program, 2009CB522300), National Natural Science Foundation of China (30830113) and Chinese Academy of Sciences (KSCX1-YW-R-24; KSCX2-YW-G-025).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Zhang, L., Shen, Y., Zhu, HJ. et al. Pentanol derivatives from basidiomycete Catathelasma imperiale and their 11β-hydroxysteroid dehydrogenases inhibitory activity. J Antibiot 62, 239–242 (2009). https://doi.org/10.1038/ja.2009.17
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2009.17
