
Abstract
Mutualism between bacteria and eukaryotes has essential roles in the history of life, but the evolution of their compatibility is poorly understood. Here we show that different Sinorhizobium strains can form either nitrogen-fixing nodules or uninfected pseudonodules on certain cultivated soybeans, while being all effective microsymbionts of some wild soybeans. However, a few well-infected nodules can be found on a commercial soybean using inocula containing a mixed pool of Tn5 insertion mutants derived from an incompatible strain. Reverse genetics and genome sequencing of compatible mutants demonstrated that inactivation of T3SS (type three secretion system) accounted for this phenotypic change. These mutations in the T3SS gene cluster were dominated by parallel transpositions of insertion sequences (ISs) other than the introduced Tn5. This genetic and phenotypic change can also be achieved in an experimental evolution scenario on a laboratory time scale using incompatible wild-type strains as inocula. The ISs acting in the adaptive evolution of Sinorhizobium strains exhibit broader phyletic and replicon distributions than other ISs, and prefer target sequences of low GC% content, a characteristic feature of symbiosis plasmid where T3SS genes are located. These findings suggest an important role of co-evolved ISs in the adaptive evolution of rhizobial compatibility.
Similar content being viewed by others


Introduction
Evolutionary concepts and analyses have an integral role in biology, affect society and promote human well-being (Losos et al., 2013; Vitti et al., 2013). To understand evolutionary processes that shape phenotypic variation, a key challenge is to reveal what portion of selection on genomes results from adaptation to particular selective agents (Losos et al., 2013), such as abiotic environment or interacting species. Bacteria are characterized by their large effective population size and short generation times and are ideal for the study of evolutionary mechanisms as demonstrated by experimental evolution of Escherichia coli pure cultures (Tenaillon et al., 2016; Lenski, 2017). However, experimental evolutionary mechanisms are largely unexplored when bacteria are experiencing a complex interaction with eukaryotic partners, such as in the pathogenic or mutualistic interactions (Kawecki et al., 2012). Despite the great diversity of bacteria, their host adaptation stages can be categorized into extracellular, facultative intracellular, obligate intracellular, obligate intracellular mutualist and organelle (Toft and Andersson, 2010). Genome reduction has been widely accepted as the major evolutionary theme of obligate intracellular symbionts, which are mainly vertically transmitted in a host-restricted manner (McCutcheon and Moran, 2012). The evolutionary processes underlying the early stages of host adaptation may be more complex due to diverse environmental stimuli and host immune pressures, to which bacteria facultatively associating with eukaryotes have to respond.
As typical facultative microsymbionts, rhizobia living saprophytically in soils can establish nitrogen-fixing nodules on root or stem of compatible legumes under nitrogen-limiting conditions, thus having a crucial role in nitrogen cycle and sustainable agriculture. The endosymbiotic form of rhizobia in legume nodules is called bacteroids, which can reduce atmospheric nitrogen into ammonia to support plant growth and obtain carbon source from plant hosts to maintain cellular activity and the energy-consuming nitrogen fixation process (Udvardi and Poole, 2013). This mutualistic rhizobium-legume symbiosis evolved around 58 million years ago (Sprent, 2007), and its establishment largely depends on rhizobial genes involved in nodulation (nod) and nitrogen fixation (nif/fix) (Remigi et al., 2016). It has been demonstrated that the horizontal transfer of key symbiosis genes (nod/nif/fix) could enable a bacterial recipient to accomplish all or part of three main steps of symbiosis establishment: nodule organogenesis, intracellular infection, and nitrogen fixation (Sullivan et al., 1995; Sullivan and Ronson, 1998; Marchetti et al., 2010; Ling et al., 2016). Transient hypermutagenesis mediated by a mutagenesis cassette co-transferred with key symbiosis genes can facilitate post-transfer adaptation of an emerging rhizobium, regarding its nodulation and intracellular infection abilities, to the new host in experimental evolution studies (Marchetti et al., 2010; Remigi et al., 2014). With these advancements of our knowledge on model rhizobial strains of large phylogenetic distances, it is hypothesized that the compatibility and performance of rhizobia on legumes need long-term evolution (Masson-Boivin et al., 2009; Remigi et al., 2016). However, we know little about the evolutionary mechanisms underlying symbiotic adaptation.
Cultivated soybean (Glycine max) was putatively domesticated from its wild progenitor Glycine soja in the region along the Yellow River of China (Li et al., 2010). Soybean nodules in this region are dominated by rhizobia belonging to Sinorhizobium differing in their symbiotic compatibility with commercial, non-commercial and wild soybeans (Wu et al., 2011; Tian et al., 2012; Guo et al., 2014; Vinardell et al., 2015; Zhang et al., 2017). Here we aimed to investigate whether certain incompatible Sinorhizobium strains have the evolutionary potential to establish compatible symbiosis with a commercial soybean grown in this region. To this end, massive screenings of Tn5 insertion mutant libraries were performed for four Sinorhizobium strains, which induce uninfected pseudonodules on an elite commercial soybean. Then reverse genetics and genome sequencing were used to verify the genetic determinants of the compatibility of those Tn5 mutants, which could form well-infected effective nodules on the test soybean cultivar. It turned out that transpositions of indigenous insertion sequences (ISs), other than the introduced Tn5, into the gene cluster encoding T3SS (type three secretion system) mainly accounted for the observed changes in symbiotic compatibility. This evolutionary potential of rhizobia was further demonstrated by inoculating wild-type strains on the same soybean cultivar and genome resequencing analysis of evolved clones. Comparative genomics and transcriptomics were further carried out to characterize these ISs acting in the adaptive evolution of rhizobial compatibility (hereafter called ‘acting ISs’). The findings were finally discussed in the context of adaptive evolution and genome ecology.
Materials and methods
Strains, plasmids, primers and growth conditions
Sinorhizobium and E. coli strains, plasmids and primers used in this work are listed in Supplementary Tables 1 and 2. Genetic procedures used for the construction of Tn5 mutant libraries, in-frame deletion mutants and site-specific insertional mutants are provided in Supplementary Materials and Methods. Culture media and the concentrations of antibiotics were described earlier (Jiao et al., 2016).
Screening Tn5 mutants and spontaneous mutants, and plant assays
The symbiotic performance of nine Sinorhizobium strains were firstly tested on two G. soja accessions (WSD and W05), the elite commercial soybean cultivar JD17 (Qin et al., 2014) grown in the region along the Yellow River, and another representative soybean cultivar C08, which is a close relative of the sequenced Williams 82 (Lam et al., 2010; Schmutz et al., 2010). Multiple independent experiments were performed.
For the screening of Tn5 mutants, which form effective nodules on JD17, a mixed pool of the Tn5 insertion mutants derived from each test strain (around 400 000 colonies for S. fredii CCBAU25509; 700 000 for S. fredii CCBAU83666; 500 000 for S. sp. III CCBAU05631; and 900 000 for S. sojae CCBAU05684) was used as inocula for 200 JD17 plants per experiment. A second independent experiment was further carried out for 05631 (around 450 000 colonies), 25509 (500 000 colonies) and 83666 (700 000 colonies). For the screening of spontaneous (SP) mutants, which are compatible with JD17, wild-type strains 05631, 25509 and 83666 were inoculated on 200 JD17 plants. All isolates forming effective nodules obtained in these screening experiments (Tn5 or SP mutants) were reinoculated on JD17 to verify their symbiotic phenotype using the high-efficient strain S. fredii CCBAU45436 as the positive control.
All seeds were surface-sterilized in 3% NaClO (wt/vol) solution and seedlings were inoculated with 1 ml of rhizobia with optical density of OD600 equivalent to 0.2. Plants were grown in vermiculite moistened with low-N nutrient solution in Leonard jars at 24° C with day/night cycles of 12/12 h. Nodules were harvested at 30 days post inoculation (dpi), and fixed with 2.5% (vol/vol) glutaraldehyde in 0.05 m cacodylate buffer when necessary (Van de Velde et al., 2006). Nodule sections were further prepared and observed using light and electron microscopies as described in Supplementary Materials and Methods.
Phylogenetic analyses
Core genomes of nine Sinorhizobium strains were determined using Bidirectional-Best-Hit method as described earlier (Tian et al., 2012). 3133 core genes were concatenated using Gblocks (Castresana, 2000) and the resulting file was used for the construction of Neighbor-Joining tree using MEGA 5 (Tamura et al., 2011).
Genome resequencing and mutation calling
The resequencing of 21 Tn5 mutants and 16 evolved clones was performed using Hiseq2000 (PE125) at SinoGenoMax (Chinese National Human Genome Center, Beijing). Bowtie 2 was used to map clean reads to the genome sequences of wild-type strains (Langmead and Salzberg, 2012). Samtools and bcftools were then used to call the genomic variations including SNPs, indels, large deletions and insertions (Li et al., 2009). The mutations identified by this procedure in each clone were further verified using PCR and Sanger sequencing. Mutations within the T3SS gene cluster were listed in Supplementary Tables 3 and 4. The genomic locations of multi-copy acting ISs were verified using PCR for representative Set_ID of ISs (Supplementary Table 5).
RNA-seq and quantitative reverse transcription PCR
Four Sinorhizobium strains were grown in yeast extract mannitol medium (YEM) at 28 °C until stationary phase (OD600≈1.2), and 3.7 μm genistein (Pérez-Montaño et al., 2016) or diluted root extract prepared as described in Supplementary Materials and Methods was supplemented when necessary. RNA samples from three independent biological replicates were collected for each treatment using the methods described earlier (Jiao et al., 2016). Samples with or without the induction of genistein were subject to RNA-seq as described earlier (Jiao et al., 2016) at SinoGenoMax. The resultant paired-end reads were mapped against the four genomes using bowtie 2 (Langmead and Salzberg, 2012). Mapped reads were counted and called using the htseq-count method and the differentially expressed genes (False discovery rate⩽0.001, |log2 ratio|⩾1) were identified by DESeq2 (Love et al., 2014; Anders et al., 2015). qRT-PCR was performed using the same procedure described in our earlier study (Jiao et al., 2016).
Statistical information
The Unweighted Pair-Group Method using arithmetic Averages (UPGMA) was used to produce the clusters of ISs based on Euclidean distance matrix of their distribution patterns on different replicons. The Normality test was carried out when applicable. F-test was used to compare the variance between the groups that are being statistically compared. All statistical tests used and described in the main text (t-test, analysis of variance (ANOVA), Tukey’s multiple comparisons, Fisher’s exact test) were two-tailed.
Data availability
RNA-seq raw data obtained in this study have been deposited in GenBank database (Bioproject no. PRJNA359737). Complete genomes of four wild-type strains used in this study have been deposited in the GenBank database (Bioproject no. PRJNA353922 and PRJNA285929).
Results
Capture of rhizobial Tn5 mutants compatible with the elite soybean cultivar
As shown in Figure 1, representative Sinorhizobium strains were generally compatible with G. soja accessions (such as WSD and W05), while certain strains formed ineffective pseudonodules on G. max cultivars JD17 and C08 (Figure 1a). In contrast to well-infected round pink nodules induced by the strain S. fredii CCBAU45436, ineffective nodules were irregular in shape and not infected by rhizobia (Figure 1b). As these Sinorhizobium strains are closely related, particularly for those strains belonging to the same species S. fredii as revealed in the phylogenomic analysis (Figure 1a), it would be interesting to know whether strains incompatible with the commercial cultivar such as G. max cv. JD17, have the potential to evolve into effective microsymbionts (Figure 2a). As evolutionary change is contingent on the initial genomic background, here four strains (25509, 83666, 05631 and 05684) representing three lineages (S. fredii, S. sojae and Sinorhizobium sp. III) of Sinorhizobium were used to test their evolutionary potential.

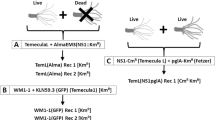
Compatible and incompatible interactions between Sinorhizobium and soybeans. (a) Neighbor-joining phylogenic tree based on 3133 core genes of Sinorhizobium strains. Bootstrap values for all nodes are 100; scale bar indicates 1% nucleotide substitution; + and –, respectively, represent compatible and incompatible interactions between test rhizobial strains and corresponding accessions of cultivated (Glycine max, JD17 and C08) and wild (Glycine soja, WSD and W05) soybeans. (b) Two micrometer thin sections (right) of JD17 nodules (left) induced by representative strains 45436 and 05631.

Capture of compatible clones by Glycine max cv. JD17. (a) Simplified model for the capture of the compatible clone by soybean plants. Pseudonodules (open circle) induced by incompatible ancestor strains (blue spot) and effective nodules (filled circle) induced by compatible clones (red spot) are shown. The abundance of compatible clones could be increased when rhizobia are released from senescent nodules. (b) Contrasting symbiotic performance of ancestor strains (05631, 83666 and 25509) and representative mutants. Transmission electron microscopy pictures of 80 nm thin section of JD17 nodules induced by these strains are shown. Red and blue arrows indicate symbiotic bacteroids and symbiosome membrane respectively (right), which are not present in sections of pseudonodules (left).
To test this possibility, a mixture of Tn5 insertion mutants of 25509 was used as inocula for 200 JD17 seedlings. As most soybean nodules were occupied by a single clone (Wang et al., 2016), this allowed JD17 to capture compatible mutants from the mutant pool in rhizosphere. Among 400 plants from two independent experiments as described in Materials and Methods, 74 pink nodules were obtained 30 dpi. Isolates from these nodules were reinoculated on JD17 and formed well-infected effective nodules (Figure 2b). When the same strategy was used for representative strains 83666, 05631 and 05684, Tn5 insertion mutants compatible with JD17 were also obtained for 83666 (297 normal nodules) and 05631 (28 normal nodules) but not for 05684 (Figure 2b), despite the fact that a larger mutant library of 05684 (around 900 000 colonies) was used in the experiment.
Mutation in T3SS is essential for rhizobial symbiotic adaptation to the elite soybean cultivar
To uncover the genetic basis of the phenotypic transition for these strains, Tn5 insertion sites were successfully determined for 122 mutants compatible with JD17 (Supplementary Table 2). Among them 112 symbiotically effective mutants harbor Tn5 insertions within genes coding for components and effector protein (NopP) of the type III secretion system (T3SS), various ABC-type transporters, transcription and signal transduction proteins, DNA helicase and ligase, reverse transcriptase, phage terminase, pyruvate carboxylase, selenium-binding protein, multiple peptide resistance factor, hypothetical proteins and so on. Eleven mutants carrying Tn5 in intergenic regions with nearby genes of diverse functions were also identified. These results raised questions whether these strains could use so many different keys to resolve the incompatibility with JD17 or are there any unidentified common contingent mutations in these isolates. To test these possibilities, we constructed pVO155 insertion mutants or deletion mutants for 77 target genes in three wild-type strains (83666, 25509 and 05631). These mutants were individually inoculated on JD17. Only mutants carrying mutations in genes encoding T3SS components, effector protein NopP or their transcriptional regulator TtsI can induce well-infected effective nodules on JD17 (Supplementary Table 2).
Genome resequencing suggests indigenous insertion sequences as efficient mutators
Consequently, we hypothesized that the mutants with Tn5 insertion in genes other than T3SS genes might harbor additional spontaneous mutation(s) in the genomic region coding for T3SS. Genome resequencing analysis of 21 randomly selected mutants (Supplementary Table 3; Figure 3a) unveiled that all of these mutants carry insertion mutations in the T3SS gene cluster mediated by either indigenous insertion sequences (ISs) (19 out of 21 mutants) or the introduced Tn5. Therefore, T3SS have a negative role in rhizobial adaptation to certain soybean cultivars as demonstrated herein and in earlier studies using both forward and reverse genetics methods (Yang et al., 2010; Tsukui et al., 2013; Tsurumaru et al., 2015). However, it remains unknown whether IS transposition is associated to Tn5 mutagenesis and whether these wild-type strains could evolve into compatible microsymbionts of JD17 without genetic manipulation, which is used in all published studies.

Characterizations of rhizobial clones compatible with G. max cv. JD17. (a) Compatible clones harbor insertion mutations in the conserved T3SS gene cluster. Sequences around individual insertion sites are conserved among three test strains, and all insertion sites are herein indicated in the T3SS gene cluster of 25509 by vertical lines (orange: 25509; light green: 83666; blue: 05631) either above (mutations in Tn5 mutants) or below (mutations in evolved clones) the DNA strands. The number in the bracket indicates the number of independent clones harboring the mutation in the same site. Tripartite regions within this T3SS cluster and genes of known functions within each region are indicated. Coding sequences on either forward (gray) or reverse (black) strand of 25509 symbiosis plasmid are shown. (b) Insertion mutation of T3SS was mainly mediated by IS (insertion sequence) among Tn5 mutants (19/21) and evolved clones with spontaneous mutations (SP; 15/16). The number of re-sequenced clones are shown. (c) The number of pink nodules on G. max cv. JD17 and G. soja WSD induced by four wild-type strains (45436, 83666, 25509 and 05631 represented in red, green, yellow and blue, respectively), 21 Tn5 mutants (05631Tn5, 83666Tn5 and 25509Tn5 mutants listed in Supplementary Table 3) and 16 SP clones (05631SP, 83666SP and 25509SP clones listed in Supplementary Table 4).
Parallel adaptive evolution of rhizobial compatibility can be achieved by spontaneous transposition of insertion sequences
To evaluate whether rhizobia are able to achieve symbiotic adaptation under the only selection pressure of JD17, wild-type 25509, 83666 and 05631 were individually inoculated on JD17. Among the inoculated 200 plants, 22, 29 and 7 pink nodules were obtained for 25509, 83666 and 05631, respectively. These nodule isolates exhibited reproducible effective symbiotic performance on JD17. Mutations in sixteen evolved clones were determined by genome resequencing (Figure 3a). Noteworthy, 15/16 evolved clones harbored IS insertions in the T3SS gene cluster and one clone had a single nucleotide insertion within the coding region of NopX, a translocator of type three effector protein (Supplementary Table 4). Moreover, it is intriguing that most mutations in the T3SS gene cluster were caused by insertion of indigenous ISs (Figure 3b) independent of whether Tn5 mutants or wild-type strains were used as inoculants. Therefore, forming uninfected pseudonodules on JD17 by these Sinorhizobium strains is due to the T3SS and the main evolutionary strategy used by them to overcome this is IS insertion.
As shown in Figure 1a, 25509, 83666 and 05631 were effective microsymbionts of G. soja but not compatible with JD17. Will the mutations in T3SS beneficial for interacting with JD17 cause symbiotic defects on G. soja? We further tested the symbiotic phenotypes of 37 re-sequenced clones on G. soja. These evolved clones were still effective microsymbionts on G. soja while being able to form effective nodules on JD17 (Figure 3c). However, this does not exclude the possibility that these evolved clones were impaired in symbiotic performance on other untested soybean accessions.
The acting insertion sequences have distinct replicon- and lineage-dependent distributions
Sequence analysis of the acting ISs in three test Sinorhizobium strains further uncovered that they belong to six distinct families (Table 1) defined in the ISfinder database (Varani et al., 2011). This reminded us of exploring the distribution of diverse ISs in genomes of Sinorhizobium strains and testing whether a pure stochastic process was involved regarding the IS recruitment.
There are totally 70, 83, 114 and 118 ISs in the genome of S. sp. III 05631, S. sojae 05684, S. fredii strains 83666 and 25509, respectively. The largest portion of IS pool was found on the symbiosis plasmid pSymA and statistically enriched herein compared with the proportion of genes harbored by this replicon (Supplementary Figure 1; Fisher’s exact test, P<0.001). The UPGMA dendrogram based on the copy number of ISs in different replicons uncovered three major clusters I, II and III (Figure 4a). Clusters I and II are enriched with those ISs, which are present on the chromosome and chromid of S. fredii strains 83666 and 25509 but absent in those of S. sp. III 05631 and S. sojae 05684 with smaller genomes (Figure 4b). Seven out of eight acting ISs belong to three subclusters (a, b and c) of III, and they show a higher conservation level across Sinorhizobium strains compared with ISs belonging to III-d, II and I (Figure 4b). Moreover, PCR analysis of genomic locations of acting ISs in representative compatible clones demonstrated that the copy number of acting ISs has been increased during adaptive evolution (Supplementary Table 5).

Acting insertion sequences in the pan-genome of rhizobia. (a) The UPGMA dendrogram of ISs based on their distribution in the tripartite genomes of Sinorhizobium strains. Family and group information of these Set_IDs are listed in Supplementary Table 6. Acting ISs are indicated in red color. The bar plot shows copy numbers of each Set_ID on three replicons depicted in different colors: orange (pSymA), green (chromid), blue (chromosome). (b) The distribution of ISs in four genomes. The red color is scaled to the relative abundance of each IS within the individual genomes. *, indicates conserved IS harbored by at least three genomes. (c) The transcription level of each set of ISs based on the sum of RPKM values within each replicon (Ch, chromosome; pB, chromid; pA, pSymA). Transcriptional profiles of four Sinorhizobium strains under the condition without the induction by genistein are pooled together herein (see Supplementary Table 6 for the full transcriptional profiles of ISs, including those obtained under the genistein induction condition). Red and blue represent the high and low expression level, respectively.
Transcriptional profiles of insertion sequences
To investigate whether these acting ISs are differentially transcribed compared with those less conserved ISs in symbiotic-like conditions, RNA-seq analysis of wild-type 83666, 25509, 05631 and 05684 was performed for free-living cultures without or with the induction by genistein, a canonical symbiosis signal secreted from soybeans (Pueppke et al., 1998). Although the nodulation genes and the T3SS genes can be actively induced by genistein, ISs did not show significant transcriptional changes (Supplementary Table 6). Moreover, certain less conserved ISs (such as, Set_ID10, Set_ID1 and Set_ID28) can be constitutively transcribed at a higher level than several acting ISs (such as Set_ID18 and Set_ID17) (Figure 4c; Supplementary Table 6). Further investigation of ISs’ transcription levels using root extracts from soybeans as inducer gave similar results as those obtained using genistein and root extracts from maize (Supplementary Figure 2). Therefore, the transcriptional upregulation of these diverse acting ISs is not an essential requirement during the adaptive evolution of rhizobial compatibility. The constitutive expression level of these ISs could be enough for the transposition activity to occur under test conditions.
Noteworthy, some transposases within the symbiosis plasmid were induced by genistein (Supplementary Table 6) and they may act in trans to promote the transposition of certain ISs (Brookfield, 2005). By summarizing transcriptional profiles of ISs and transposases (Supplementary Table 6) identified by ISfinder and the annotation pipeline of RAST (Varani et al., 2011; Overbeek et al., 2014), we found that S. sojae 05684 harbored significantly less number of transcribed ISs and transposases in the context of the pan-genome of four Sinorhizobium strains (Figure 5a; Fisher’s exact test, P<0.001). This might be the reason why we failed to obtain the evolved clones, which are compatible with JD17, from 05684 under the test conditions. Indeed, reverse genetics confirmed that the T3SS mutant of 05684 can form effective nitrogen-fixing nodules on JD17 (Supplementary Table 2). Therefore, S. sojae 05684 seems to have a lower evolutionary potential compared with the other three Sinorhizobium strains.

Distribution of transcribed ISs and acting ISs. (a) The strain-dependent distribution of transcribed ISs (identified by ISfinder) and transposases (annotated by RAST software). (b) GC% of sequences surrounding acting ISs and replicon-specific clusters of ISs. Statistically significant difference is indicated (*P<0.05; **P<0.01; ***P<0.001) based on either Tukey’s multiple comparisons (b) or the Fisher’s exact test (a). Error bar represents s.e.m.
The acting ISs prefer target sequences of lower GC%
The T3SS genes are also located on pSymA, which is characterized not only by harboring the key symbiosis genes but also by its lower GC% in the Sinorhizobium pan-genome: 58.89–59.34% compared with 62.44–62.88% of chromosome and 61.90–62.89% of chromid. Intriguingly, GC% of the surrounding sequences of acting ISs is lower than that of chromosome-specific ISs (Figure 5b): 54.74±1.09% (mean±s.e.m.) compared with 62.46±1.42% (cluster I; Tukey’s multiple comparisons, P<0.05). The latter value is significantly higher than those of pSymA-specific ISs (cluster III, 54.88±1.12% Tukey’s multiple comparisons, P<0.05) and chromid-specific ISs (cluster II, 56.20±1.95% Tukey’s multiple comparisons, P=0.0606). Despite these differences in GC% of surrounding sequences, ISs’ own sequences did not show significant variations among different clusters (Supplementary Figure 3).
Discussion
In this study, we demonstrate a fast adaptive evolution of rhizobial compatibility without artificial genetic manipulation, that is, parallel transpositions of a preferred subset of indigenous ISs into genes, encoding T3SS apparatus components, the effector protein NopP, and their transcriptional activator TtsI, can enable incompatible Sinorhizobium strains to induce well-infected effective nodules on an elite soybean cultivar JD17.
The observed negative impact of Sinorhizobium T3SS genes on nodulating certain soybean cultivar is consistent with earlier reverse genetics studies. For example, the inactivation of nopP led to an increase in the symbiotic capacity of S. fredii HH103 on Williams soybean and the presence of T3SS can restrict the symbiosis of S. fredii USDA257 on certain genotypes of soybeans (López-Baena et al., 2009; Yang et al., 2010). It has been demonstrated that most components within the T3SS gene cluster can exert positive, negative or neutral effects on the establishment of symbiosis depending on the genetic background of diverse rhizobia and their legume hosts (Skorpil et al., 2005; López-Baena et al., 2009; Yang et al., 2010; Staehelin and Krishnan, 2015; Okazaki et al., 2015). The system-dependent role of T3SS in symbiosis has also been demonstrated in Bradyrhizobium elkanii USDA61, which uses T3SS to activate host nodulation signaling by bypassing Nod factor recognition in soybean roots (Okazaki et al., 2013).
Our findings further highlight that a preferred subset of indigenous ISs can efficiently inactivate T3SS during adaptive evolution of Sinorhizobium compatibility. This supports the view that transposable elements should be no longer considered as ‘junk’ and ‘selfish’ components in genomes but an important player in evolution (Biémont, 2010). Similarly, the transposable phage Φ4 accelerated the loss of clinically important virulence-related traits of Pseudomonas aeruginosa, mediating and driving parallel adaptive evolution in pathogen biofilms (Davies et al., 2016). The acting ISs revealed in this study exhibited a higher efficiency in insertion mutation within the T3SS region than introduced Tn5 and spontaneous point mutation, at least partially due to their high-copy numbers and enrichment in the symbiosis plasmid where the T3SS cluster is located. This is in contrast to earlier report that point mutation caused by the hypermutagenesis cassette is the key mechanism underlying the experimental evolution of the genetically engineered Ralstonia solanacearum into a novel microsymbiont of Mimosa (Remigi et al., 2014), and is distinct from the adaptation of free-living bacteria such as E. coli through accumulating beneficial point mutations in the long-term experimental evolution (Tenaillon et al., 2016).
Sinorhizobium strains are characterized by their tripartite genomes (chromosome, the chromid pSymB, and the symbiosis plasmid pSymA), which underwent replicon-dependent evolutionary histories (Galardini et al., 2013; Guo et al., 2014). Here we identified chromosome- and chromid-specific clusters (I and II) of ISs, the expansion of which in S. fredii strains compared with S. sojae and S. sp. III might be associated with the speciation process of these species and contribute to the larger genomes of S. fredii (Tian et al., 2012). These findings are in line with the increasing evidence that differences in transposable element content make substantial contribution to diversity in genome size (Linquist et al., 2013). Moreover, comparative genomics and metabolic modeling evidences suggest that the chromid may have a critical role in the growth of Sinorhizobium meliloti in bulk soils and rhizosphere, and contribute substantially to strain differentiation (Galardini et al., 2013; diCenzo et al., 2016). Previous investigations of rhizobial diversity in both nodules and rhizosphere soils of soybeans across eco-regions demonstrated that the distribution of S. sojae and S. sp. III was geographically limited (Tian et al., 2012; Zhang et al., 2017), indicating their restricted adaptability. It was further uncovered herein that S. sojae 05684 showing the lower potential to evolve into compatible microsymbionts of the elite soybean cultivar JD17, at least partially due to the lower number of transcribed ISs and transposases under the same conditions used for the other three test strains.
The chromosome and chromid are generally subject to intraspecific microevolutionary processes and determine the vertical evolutionary history of rhizobial species (selfing), while the symbiosis plasmid harboring the symbiosis island is well known for its horizontal transfer ability among species (outcrossing) (Harrison et al., 2010; Guo et al., 2014; Remigi et al., 2016). Herein we uncovered that the ‘outcrossing’ pSymA contained more ISs than the relatively ‘selfing’ chromosome and chromid. This phenomenon to certain extent mimics the findings in eukaryotes such as the outcrossing Arabidopsis lyrata harboring more transposable elements than the selfing plant Arabidopsis thaliana, and the sexual but not asexual strains of yeast showing rapid spread of a novel foreign transposon (Zeyl et al., 1996; Hu et al., 2011; Agren, 2014). Although a bacterial recipient may benefit from the horizontally acquired (or infected) symbiosis plasmid in the long run (Remigi et al., 2016), a stable bacteria-plasmid association needs an amelioration of the cost-of-carriage through modulating the activity of excess accessory genes within the plasmid (Harrison and Brockhurst, 2012). This is particularly important for rhizobia, which experience fluctuating soil conditions and diverse germplasms of legume hosts. The enrichment of ISs on pSymA could be postulated as an effective evolutionary strategy during the co-evolution of this symbiosis plasmid and its host.
Indeed, the ISs acting in the adaptive evolution of Sinorhizobium compatibility are enriched on pSymA. It is interesting to further investigate whether ISs on the ‘outcrossing’ pSymA are usually more active than ISs from ‘selfing’ replicons under different conditions. Noteworthy, prophages also characterized by their infectious route of transmission are active regulatory switches of crucial bacterial processes such as virulence and stress resistance (Feiner et al., 2015; Davies et al., 2016). Moreover, these acting ISs are characterized by their wider phyletic and replicon distribution patterns compared with those ISs only found on pSymA (the subcluster III-d) and those enriched on the chromosome (cluster I) and chromid (cluster II). These findings may fit well to the framework of the ecology of genome where transposable elements including ISs are analogous to species in ecological communities (Brookfield, 2005; Venner et al., 2009). This view is supported by several lines of evidence obtained in this study. Firstly, the acting ISs seem to prefer targets of lower GC% level, supporting the hypothesis that GC% could be an important genetic parameter shaping the distribution of diverse ISs in the context of genome ecology (Linquist et al., 2013). Secondly, the increased copy number of acting ISs during adaptive evolution (Supplementary Table 5) is in line with the view that to increase its own population size is the dream of any ‘species’. Thirdly, the enrichment of these acting ISs in the horizontally transferable symbiosis plasmid (Pérez-Mendoza et al., 2005; Epstein et al., 2012; Guo et al., 2014) could allow their dispersal in the pan-genome of related Sinorhizobium species.
It is considered that a conserved element in (pan-)genome is very likely to perform a function that is adaptive for the host (Brookfield, 2005; Feiner et al., 2015). In this study, rhizobial strains benefited from the transposition of conserved ISs during the adaptive evolution of symbiotic compatibility. The higher evolvability associated with these acting ISs than introduced Tn5 and spontaneous point mutation can be considered as a beneficial trait for rhizobia as demonstrated earlier for the mutagenesis imuABC cassette co-transferred with symbiosis genes (Remigi et al., 2014), and thus the presence of these conserved acting ISs might be favored by a selection process shared by test Sinorhizobium spp. Indeed, all of them can nodulate soybeans, and key symbiosis genes and T3SS genes are located on the symbiosis plasmid where co-evolved acting ISs are enriched.
Conclusion
Significant advances have been made in the molecular mechanisms underlying the transposition of transposable elements (Mcclintock, 2016), and here we further demonstrated that a preferred subset of ISs can make substantial contributions to the adaptive evolution of rhizobial compatibility on a laboratory time scale. These acting ISs are ecologically successful by occupying more lineages and replicons in the pan-genome of test rhizobial strains sharing legume hosts. They prefer target sequences of low GC% content, a characteristic feature of symbiosis plasmid where key symbiosis genes and T3SS genes are located. These acting ISs can be postulated as ‘species’ co-evolved with their hosts in the context of genome ecology. These findings are not only important for our understanding of the evolution of interactions between bacteria (symbiotic or pathogenic) and eukaryotes, but also shedding new light on the way we could study the general theory of genome ecology.
References
Agren JA . (2014). Evolutionary transitions in individuality: insights from transposable elements. Trends Ecol Evol 29: 90–96.
Anders S, Pyl PT, Huber W . (2015). HTSeq A Python framework to work with high-throughput sequencing data. Bioinformatics 31: 166–169.
Biémont C . (2010). A brief history of the status of transposable elements: From junk DNA to major players in evolution. Genetics 186: 1085–1093.
Brookfield JFY . (2005). The ecology of the genome-mobile DNA elements and their hosts. Nat Rev Genet 6: 128–136.
Castresana J . (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 17: 540–552.
Davies EV, James CE, Williams D, O’Brien S, Fothergill JL, Haldenby S et al. (2016). Temperate phages both mediate and drive adaptive evolution in pathogen biofilms. Proc Natl Acad Sci USA 113: 8266–8271.
diCenzo GC, Checcucci A, Bazzicalupo M, Mengoni A, Viti C, Dziewit L et al. (2016). Metabolic modelling reveals the specialization of secondary replicons for niche adaptation in Sinorhizobium meliloti. Nat Commun 7: 1–10.
Epstein B, Branca A, Mudge J, Bharti AK, Briskine R, Farmer AD et al. (2012). Population genomics of the facultatively mutualistic bacteria Sinorhizobium meliloti and S. medicae. PLoS Genet 8: e1002868.
Feiner R, Argov T, Rabinovich L, Sigal N, Borovok I, Herskovits AA . (2015). A new perspective on lysogeny: prophages as active regulatory switches of bacteria. Nat Rev Microbiol 13: 641–650.
Galardini M, Pini F, Bazzicalupo M, Biondi EG, Mengoni A . (2013). Replicon-dependent bacterial genome evolution: the case of Sinorhizobium meliloti. Genome Biol Evol 5: 542–558.
Guo HJ, Wang ET, Zhang XX, Li QQ, Zhang YM, Tian CF et al. (2014). Replicon-dependent differentiation of symbiosis-related genes in Sinorhizobium nodulating Glycine max. Appl Environ Microbiol 80: 1245–1255.
Harrison E, Brockhurst MA . (2012). Plasmid-mediated horizontal gene transfer is a coevolutionary process. Trends Microbiol 20: 262–267.
Harrison PW, Lower RPJ, Kim NKD, Young JPW . (2010). Introducing the bacterial ‘chromid’: not a chromosome, not a plasmid. Trends Microbiol 18: 141–148.
Hu TT, Pattyn P, Bakker EG, Cao J, Cheng J, Richard M et al. (2011). The Arabidopsis lyrata genome sequence and the basis of rapid genome size change. Nat Genet 43: 476–481.
Jiao J, Wu LJ, Zhang B, Hu Y, Li Y, Zhang XX et al. (2016). MucR is required for transcriptional activation of conserved ion transporters to support nitrogen fixation of Sinorhizobium fredii in soybean nodules. Mol Plant-Microbe Interact 29: 352–361.
Kawecki TJ, Lenski RE, Ebert D, Hollis B, Olivieri I, Whitlock MC . (2012). Experimental evolution. Trends Ecol Evol 27: 547–560.
Lam H-M, Xu X, Liu X, Chen W, Yang G, Wong F-L et al. (2010). Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection. Nat Genet 42: 1053–1059.
Langmead B, Salzberg SL . (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359.
Lenski RE . (2017). What is adaptation by natural selection? Perspectives of an experimental microbiologist. PLoS Genet 13: e1006668.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25: 2078–2079.
Li Y-H, Li W, Zhang C, Yang L, Chang R-Z, Gaut BS et al. (2010). Genetic diversity in domesticated soybean (Glycine max and its wild progenitor (Glycine soja for simple sequence repeat and single-nucleotide polymorphism loci. New Phytol 188: 242–253.
Ling J, Wang H, Wu P, Li T, Tang Y, Naseer N et al. (2016). Plant nodulation inducers enhance horizontal gene transfer of Azorhizobium caulinodans symbiosis island. Proc Natl Acad Sci USA 113: 13875–13880.
Linquist S, Saylor B, Cottenie K, Elliott TA, Kremer SC, Gregory TR . (2013). Distinguishing ecological from evolutionary approaches to transposable elements. Biol Rev 575: 573–584.
López-Baena FJ, Monreal JA, Pérez-Montaño F, Guasch-Vidal B, Bellogín RA, Vinardell JM et al. (2009). The absence of Nops secretion in Sinorhizobium fredii HH103 increases GmPR1 expression in Williams soybean. Mol Plant-Microbe Interact 22: 1445–1454.
Losos JB, Arnold SJ, Bejerano G, Brodie ED, Hibbett D, Hoekstra HE et al. (2013). Evolutionary biology for the 21st century. PLoS Biol 11: e1001466.
Love MI, Huber W, Anders S . (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550.
Marchetti M, Capela D, Glew M, Cruveiller S, Chane-Woon-Ming B, Gris C et al. (2010). Experimental evolution of a plant pathogen into a legume symbiont. PLoS Biol 8: e1000280.
Masson-Boivin C, Giraud E, Perret X, Batut J . (2009). Establishing nitrogen-fixing symbiosis with legumes: how many rhizobium recipes? Trends Microbiol 17: 458–466.
Mcclintock B . (2016). Barbara McClintock on defining the unstable genome. Genetics 204: 3–4.
McCutcheon JP, Moran NA . (2012). Extreme genome reduction in symbiotic bacteria. Nat Rev Microbiol 10: 13–26.
Okazaki S, Kaneko T, Sato S, Saeki K . (2013). Hijacking of leguminous nodulation signaling by the rhizobial type III secretion system. Proc Natl Acad Sci USA 110: 17131–17136.
Okazaki S, Tittabutr P, Teulet A, Thouin J, Fardoux J, Chaintreuil C et al. (2015). Rhizobium – legume symbiosis in the absence of Nod factors: two possible scenarios with or without the T3SS. ISME J 10: 64–74.
Overbeek R, Olson R, Pusch GD, Olsen GJ, Davis JJ, Disz T et al. (2014). The SEED and the rapid annotation of microbial genomes using subsystems Technology (RAST). Nucleic Acids Res 42: 206–214.
Pérez-Mendoza D, Sepúlveda E, Pando V, Muñoz S, Nogales J, Olivares J et al. (2005). Identification of the rctA gene, which is required for repression of conjugative transfer of rhizobial symbiotic megaplasmids. J Bacteriol 187: 7341–7350.
Pérez-Montaño F, Jiménez-Guerrero I, Acosta-Jurado S, Navarro-Gómez P, Ollero FJ, Ruiz-Sainz JE et al. (2016). A transcriptomic analysis of the effect of genistein on Sinorhizobium fredii HH103 reveals novel rhizobial genes putatively involved in symbiosis. Sci Rep 6: 31592.
Pueppke SG, Bolanos-Vasquez MC, Werner D, Bec-Ferte MP, Prome JC, Krishnan HB . (1998). Release of flavonoids by the soybean cultivars McCall and Peking and their perception as signals by the nitrogen-fixing symbiont Sinorhizobium fredii. Plant Physiol 117: 599–608.
Qin J, Wang F, Gu F, Wang J, Chen Q, Zhang M . (2014). A genetic composition analysis of soybean sibling varieties Jidou17 and Ji nf58. Aust J Crop Sci 8: 791–798.
Remigi P, Capela D, Clerissi C, Tasse L, Torchet R, Bouchez O et al. (2014). Transient hypermutagenesis accelerates the evolution of legume endosymbionts following horizontal gene transfer. PLOS Biol 12: e1001942.
Remigi P, Zhu J, Young JPW, Masson-Boivin C . (2016). Symbiosis within symbiosis: evolving nitrogen-fixing legume symbionts. Trends Microbiol 24: 63–75.
Schmutz J, Cannon SB, Schlueter J, Ma J, Mitros T, Nelson W et al. (2010). Genome sequence of the palaeopolyploid soybean. Nature 463: 178–183.
Skorpil P, Saad MM, Boukli NM, Kobayashi H, Ares-Orpel F, Broughton WJ et al. (2005). NopP, a phosphorylated effector of Rhizobium sp strain NGR234, is a major determinant of nodulation of the tropical legumes Flemingia congesta and Tephrosia vogelii. Mol Microbiol 57: 1304–1317.
Sprent JI . (2007). Evolving ideas of legume evolution and diversity: a taxonomic perspective on the occurrence of nodulation. New Phytol 174: 11–25.
Staehelin C, Krishnan HB . (2015). Nodulation outer proteins: double-edged swords of symbiotic rhizobia. Biochem J 470: 263–274.
Sullivan JT, Patrick HN, Lowther WL, Scott DB, Ronson CW . (1995). Nodulating strains of Rhizobium loti arise through chromosomal symbiotic gene transfer in the environment. Proc Natl Acad Sci USA 92: 8985–8989.
Sullivan JT, Ronson CW . (1998). Evolution of rhizobia by acquisition of a 500- kb symbiosis island that integrates into a phe-tRNA gene. Proc Natl Acad Sci USA 95: 5145–5149.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S . (2011). MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28: 2731–2739.
Tenaillon O, Barrick JE, Ribeck N, Deatherage DE, Blanchard JL, Dasgupta A et al. (2016). Tempo and mode of genome evolution in a 50,000 - generation experiment. Nature 536: 165–170.
Tian CF, Zhou YJ, Zhang YM, Li QQ, Zhang YZ, Li DF et al. (2012). Comparative genomics of rhizobia nodulating soybean suggests extensive recruitment of lineage-specific genes in adaptations. Proc Natl Acad Sci USA 109: 8629–8634.
Toft C, Andersson SGE . (2010). Evolutionary microbial genomics: insights into bacterial host adaptation. Nat Rev Genet 11: 465–475.
Tsukui T, Eda S, Kaneko T, Sato S, Okazaki S, Kakizaki-chiba K et al. (2013). The type III secretion system of Bradyrhizobium japonicum USDA122 mediates symbiotic incompatibility with Rj2 soybean. Appl Environ Microbiol 79: 1048–1051.
Tsurumaru H, Hashimoto S, Okizaki K, Kanesaki Y, Yoshikawa H, Yamakawa T . (2015). A putative type III secretion system effector encoded by the MA20_12780 gene in Bradyrhizobium japonicum Is-34 causes incompatibility with Rj4 genotype soybeans. Appl Environ Microbiol 81: 5812–5819.
Udvardi M, Poole PS . (2013). Transport and metabolism in legume-rhizobia symbioses. Annu Rev Plant Biol 64: 781–805.
Varani AM, Siguier P, Gourbeyre E, Charneau V, Chandler M . (2011). ISsaga is an ensemble of web-based methods for high throughput identification and semi-automatic annotation of insertion sequences in prokaryotic genomes. Genome Biol 12: R30.
Van de Velde W, Guerra JCP, de Keyser A, de Rycke R, Rombauts S, Maunoury N et al. (2006). Aging in legume symbiosis. A molecular view on nodule senescence in Medicago truncatula. Plant Physiol 141: 711–720.
Venner S, Feschotte C, Biémont C . (2009). Dynamics of transposable elements: towards a community ecology of the genome. Trends Genet 25: 317–323.
Vinardell J-M, Acosta-Jurado S, Zehner S, Göttfert M, Becker A, Baena I et al. (2015). The Sinorhizobium fredii HH103 genome: A comparative analysis with S. fredii strains differing in their symbiotic behavior with soybean. Mol Plant-Microbe Interact 28: 811–824.
Vitti JJ, Grossman SR, Sabeti PC . (2013). Detecting natural selection in genomic data. Annu Rev Genet 47: 97–120.
Wang D, Wang YC, Wu LJ, Liu JX, Zhang P, Jiao J et al. (2016). Construction and pilot screening of a signature-tagged mutant library of Sinorhizobium fredii. Arch Microbiol 198: 91–99.
Wu LJ, Wang HQ, Wang ET, Chen WX, Tian CF, Chen WX . (2011). Genetic diversity of nodulating and non-nodulating rhizobia associated with wild soybean (Glycine soja Sieb. & Zucc.) in different ecoregions of China. FEMS Microbiol Ecol 76: 439–450.
Yang S, Tang F, Gao M, Krishnan HB, Zhu H . (2010). R gene-controlled host specificity in the legume-rhizobia symbiosis. Proc Natl Acad Sci USA 107: 18735–18740.
Zeyl C, Bell G, Green DM . (1996). Sex and the spread of retrotransposon Ty3 in experimental populations of Saccharomyces cerevisiae. Genetics 143: 1567–1577.
Zhang XX, Guo HJ, Jiao J, Zhang P, Xiong HY, Chen WX et al. (2017). Pyrosequencing of rpoB uncovers a significant biogeographical pattern of rhizobial species in soybean rhizosphere. J Biogeogr 44: 1491–1499.
Acknowledgements
We thank Meng Ni and Honming Lam from the Chinese University of Hong Kong for assistance in closing genome gaps of CCBAU25509. This work was supported by the National Basic Research Program of China (973 program 2015CB158300), the National Natural Science Foundation of China (31522003) and the Innovative Project of State Key Laboratory of Agrobiotechnology (2014SKLAB4-1).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Zhao, R., Liu, L., Zhang, Y. et al. Adaptive evolution of rhizobial symbiotic compatibility mediated by co-evolved insertion sequences. ISME J 12, 101–111 (2018). https://doi.org/10.1038/ismej.2017.136
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2017.136
This article is cited by
-
Symbiotic efficiency of Rhizobium leguminosarum sv. trifolii strains originating from the subpolar and temperate climate regions
Scientific Reports (2024)
-
Intracellular common gardens reveal niche differentiation in transposable element community during bacterial adaptive evolution
The ISME Journal (2023)
-
Rhizobial migration toward roots mediated by FadL-ExoFQP modulation of extracellular long-chain AHLs
The ISME Journal (2023)
-
The zinc-finger bearing xenogeneic silencer MucR in α-proteobacteria balances adaptation and regulatory integrity
The ISME Journal (2022)
-
Evolution of rhizobial symbiosis islands through insertion sequence-mediated deletion and duplication
The ISME Journal (2022)
