
Abstract
Soils of northern temperate and boreal forests represent a large terrestrial carbon (C) sink. The fate of this C under elevated atmospheric CO2 and climate change is still uncertain. A fundamental knowledge gap is the extent to which ectomycorrhizal fungi (EMF) and saprotrophic fungi contribute to C cycling in the systems by soil organic matter (SOM) decomposition. In this study, we used a novel approach to generate and compare enzymatically active EMF hyphae-dominated and saprotrophic hyphae-enriched communities under field conditions. Fermentation-humus (FH)-filled mesh bags, surrounded by a sand barrier, effectively trapped EMF hyphae with a community structure comparable to that found in the surrounding FH layer, at both trophic and taxonomic levels. In contrast, over half the sequences from mesh bags with no sand barrier were identified as belonging to saprotrophic fungi. The EMF hyphae-dominated systems exhibited levels of hydrolytic and oxidative enzyme activities that were comparable to or higher than saprotroph-enriched systems. The enzymes assayed included those associated with both labile and recalcitrant SOM degradation. Our study shows that EMF hyphae are likely important contributors to current SOM turnover in sub-boreal systems. Our results also suggest that any increased EMF biomass that might result from higher below-ground C allocation by trees would not suppress C fluxes from sub-boreal soils.
Similar content being viewed by others

Introduction
Northern temperate and boreal forests represent a large carbon (C) sink, with an estimated half a gigatonne of C sequestered in above ground biomass every year (Myneni et al., 2001). Much of this C is eventually transferred below ground, either after the death or annual senescence of vegetation or directly via photosynthetic transfer to root systems and associated ectomycorrhizal fungi (EMF). These soils store up to three times the amount of C as is stored in the above ground vegetation, primarily in the form of soil organic matter (SOM) (Schmidt et al., 2011). These C sinks have the potential to become C sources under the projected global climate change (IPCC, 2007). Although some climate models assume increased C storage in temperate forest soils because of the increased plant allocation of photosynthetic C to roots and symbiotic fungi (Drigo et al., 2010; Orwin et al., 2011; Clemmensen et al., 2013), there is a high degree of uncertainly in this assumption (Higgins and Harte, 2012). Increased C allocation with elevated CO2 may not only be short-lived (Norby et al., 2010), but concomitant increases in mycorrhizal and mycorrhizosphere activity may stimulate decomposition of previously recalcitrant SOM (Cheng et al., 2012; Phillips et al., 2012b). Fundamental gaps in our understanding of terrestrial C cycling, including the relative contributions of EMF and other fungi to the release of SOM-C (Bargett, 2011), limit the predictive capability of current models (Von Lützow and Kögel-Knabner, 2009; Hayes et al., 2012).
Fungi, especially saprotrophic fungi, are thought to dominate organic matter turnover in temperate forest soils (Baldrian, 2008). Certain groups of saprotrophic fungi, specifically the white rot fungi, have unique abilities to degrade wood because they possess numerous copies of class II peroxidases (Floudas et al., 2012). Both saprotrophic and EMF species, however, produce a range of hydrolytic and oxidative enzymes that have the potential to break down C-containing compounds and mobilize nutrients from SOM (Courty et al., 2010; Floudas et al., 2012). Although photosynthates are likely the primary source of C used by EMF under ideal conditions (Treseder et al., 2006; Wolfe et al., 2012), current research suggests that these fungi may also directly (Vaario et al., 2012) or indirectly (Rineau et al., 2012) access SOM-C pools.
Recalcitrant SOM, including lignin and humic complexes, is predominantly found in soil horizons dominated by EMF mycelial networks (Lindahl et al., 2007). These mycelia are present at up to 600 kg of hyphae per hectare (Wallander et al., 2001; Hendricks et al., 2006) and account for up to one-third of the total microbial biomass in coniferous forests (Högberg and Högberg, 2002; Cairney, 2012). Although EMF mats are known to increase total soil CO2 respiration (Phillips et al., 2012a), the full contribution of these fungi, and their extensive mycelia, to decomposition processes remains unknown (van der Wal et al., 2012). Given the current interest in the C sequestration potential of boreal and temperate forests (De Luca and Boisvenue, 2012), as well as the influence of any increased C allocation below ground by plants, the role of EMF mycelia in SOM-C turnover warrants increased investigation (Talbot et al., 2008; Ekblad et al., 2013).
Current research to estimate the extent and activity of EMF mycelia rely on methods to exclude roots and most saprotrophic fungi from a given zone in the soil profile. Hyphal in-growth cores or bags are commonly used (Wallander et al., 2001). These hyphae traps are usually filled with an inert substrate, such as sand, that minimizes colonization by saprotrophic fungi (Korkama et al., 2007; Lindahl et al., 2007; Wallander et al., 2010). In some cases, additional nutrient sources are added to ensure sufficient hyphal colonization or to evaluate the impact of limiting nutrients on EMF biomass production (Hagerberg and Wallander, 2002; Hagerberg et al., 2003; Hedh et al., 2008; Potila et al., 2009; Berner et al., 2012). However, the captured extramatrical hyphae may not accurately represent the composition or abundance of EMF communities in the surrounding soil (Hendricks et al., 2006; Kjøller, 2006). Further, enzyme expression and activity is downregulated when no nutrients are present (Wright et al., 2005). In order to estimate the contribution of EMF to the terrestrial C cycle, the potential SOM-degrading enzyme activity in locally abundant EMF hyphae needs to be determined.
To investigate the relative contribution of EMF mycelia to enzyme activities in a sub-boreal spruce forest, we buried mesh bags filled with sterilized organic material with or without a surrounding sand barrier. Bags with a sand barrier were served to trap enzymatically active EMF hyphae while excluding obligately saprotrophic hyphae. Filling the mesh bags with fermentation-humic (FH) substrate taken from the same soil in which the traps were to be buried enabled a direct comparison between the EMF hyphae in the mesh bags and the surrounding FH layer. Based on our previous research, which showed that EMF root tips in these ecosystems had high hydrolytic and oxidative enzyme activities (Jones et al., 2010, 2012), we hypothesized that EMF-dominated communities would have a SOM-degradation potential comparable to that of saprotroph-dominated communities. High-throughput sequencing, which combined with comprehensive measures of enzymes involved in SOM turnover, allowed us to evaluate the relative abundance and structure of fungal communities and to elucidate the potential role of EMF fungi in the soil C cycle.
Materials and methods
Field site
The field site was part of a British Columbia Ministry of Forests study that investigated the impact of fertilization on forest productivity and below-ground ecosystem processes (Brockley and Simpson, 2004). The current study occurred at the Crow Creek site, established in 1994 near Houston, British Columbia, Australia (54° 20′ 126° 17′) in the sub-boreal spruce biogeoclimatic zone, on Eluviated Dystric Brunisol to Orthic Humo-Ferric Podzol soils. Each treatment within the site consisted 0.164 hectare plots replicated three times. The current study utilized the non-fertilized control and two fertilized (annually and every 6 years) treatments; for further site information refer to Brockley and Simpson (2004). During the season of this study, the FH layer averaged 33% total C, 1.06% total N, 64% organic matter, NH4-N, NO3-N and available P of 46, 2.9 and 346 mg kg−1, respectively, and pH(CaCl2) 4.35 (British Columbia Ministry of Forests Research Laboratory, Victoria, British Columbia, Australia).
Mesh bag construction for EMF extramatrical hyphal capture
FH layer substrate was collected from each treatment plot in the fall of 2008. Ten random sub-samples per plot were composited, sieved (4.5 mm) and stored at 4 °C until use. Eleven hyphal trap bags (4.5 × 3.5 cm) were constructed for each treatment plot (99 bags in total) using 50 μm mesh (NITEX 03-50/31; Sefar Inc., Depew, NY, USA). Each bag was filled with 15 g FH substrate, heat-sealed and then sterilized by gamma irradiation with a total dose of 6 Mrad (2 × 3 Mrad with a 1 week resting interval). Post irradiation, bags were stored at 4 °C for an 8-week stabilization period to minimize residual enzyme activity (Lensi et al., 1991). Substrate from one bag from each plot was serially diluted in monopotassium phosphate buffer, plated in triplicate on modified Melin–Norkrans or tryptic soy agar plates (Phillips et al., 2006) and incubated for 1 month to assess for fungal or bacterial contamination, respectively. No bacterial or fungal growth was observed on culture plates after 1 month of incubation, indicating that two consecutive doses of irradiation effectively eliminated residual spores. Irradiation did not significantly change the pH of the substrates. Another sub-sample of these controls was assessed for residual enzyme activity and pH. Bags intended to capture EMF but exclude saprotrophic hyphae (n=5 per plot) were placed within sand-filled plastic containers (NetPak, Coquitlam, British Columbia, Australia). These containers were punched with circular holes (6 mm diameter; 28 holes per horizontal face and 10 holes per vertical face) to allow hyphal access, lined with a 50-μm NITEX mesh and filled to a depth of 1 cm with sterile silica sand (HCl acid washed and autoclaved). Substrate-containing mesh bags were placed on the sand, covered with 1 cm additional sand and then a final top layer of mesh was sealed to the initial liner using silicone sealant to form a continuous mesh envelope (Supplementary Figure 1). Bags intended to capture both EMF and saprotrophic hyphae (n=5 each plot) were also encapsulated within another layer of mesh, but had no intervening sand barrier.
Hyphal trap bags were buried at the Crow Creek site on 5 June 2009. Each set of bags (with or without sand barriers, +SB and –SB, respectively) were placed in the FH layer of the treatment plots from which the substrates were originally taken. One pair of +SB and –SB bags was buried at five random spots within each plot, 60 cm from the base of a tree and 60 cm from each other. All bags and an FH sample (10 × 10 cm) from between each set were harvested on 1 October 2009. Upon return to the lab, the five +SB, −SB or FH sub-samples from individual plots were composited, so that a single representative +SB, −SB and FH sample was obtained per plot. Sand from the +S mesh bags was also composited on a plot basis. Half of each composited sample was frozen at −20 °C for subsequent molecular analysis and half was stored at 4 °C overnight for use in enzyme assays.
Enzyme assays
All +SB, −SB, FH, sand and residual-activity control samples, as well as EMF tips selected from the FH samples, were assessed for their organic matter breakdown potential. The assessed enzymes included β-1,4-glucosidase (EC 3.2.1.21), cellobiohydrolase (EC 3.2.1.91), β-1,4-xylosidase (EC 3.2.1.37), peroxidase (EC 1.11.1.7), laccase (EC 1.10.3.2), phenol oxidase (EC 1.14.18.1 and EC 1.10.3.2), β-1,4-N-acetylglucosaminidase (EC 3.2.1.52), acid phosphatase (EC 3.1.3.2), leucine aminopeptidase (EC 3.4.11.1) and sulfatase (EC 3.1.6.1). Note that although laccases are phenol oxidases, the term laccase is used in the study to differentiate a 2,2′-azino-bis(3-ethylbenzothiazoline-6 sulfonic acid) diammonium salt (ABTS) assay from a L-3, 4-dihydroxyphenylalanine (L-DOPA) assay. Full details of assay conditions are provided in Supplementary Table S1. All assays except laccase, peroxidase and phenol oxidase were fluorometric.
For +SB, −SB, FH and residual-activity control assays, 5 g of sample was homogenized with a sterile mortar and pestle, 0.1 g of sub-sample was transferred to a sterile Nalgene bottle containing 100 ml of 50 mM sodium acetate buffer (pH 5.0), and then suspensions were shaken for 1 h at 500 r.p.m. Sand samples were similarly processed using 10 g of sample and 1 g of sub-sample. Fluorometric and colorimetric assays were then performed according to previously published methods (Saiya-Cork et al., 2002 and Sinsabaugh et al., 2003), fully described in the Supplementary Information.
For EMF tip assays, roots from the FH sample were gently washed, separated under a dissecting microscope, and ∼7 turgid EMF tips from each of the four most common observed morphotypes were randomly selected from each sample. Tips were cleaned of debris and placed in individual microsieves in 96-well microplates containing 75 mM Tris-maleic acid buffer (pH 4.5) to equilibrate for at least 5 min before beginning the sequential enzyme assays (Supplementary Table S1). Fluorometric and colorimetric assays were then performed according to previously published methods (Courty et al., 2005 and Pritsch et al., 2004), fully described in the Supplementary Information. After the final assay, tips were removed from the sieves and scanned (Scanmaker 8700; Microtek Lab, Sante Fe Springs, CA, USA), and their surface areas were determined (WinRHIZO, Regent Instruments, Québec, QC, Canada). Tips were then frozen at −80 °C for subsequent molecular analysis. The total enzymatic activity of EMF tips per plot was determined by averaging the activities of all tips. EMF taxon-specific activities were determined by averaging the activity of the (up to) seven EMF tips per plot, whose identities were confirmed by morphological and molecular identification.
Identification of fungal communities
Sequencing of ectomycorrhizal tips
DNA was extracted from EMF tips using the Sigma Extract-N-Amp Plant Kit (Sigma-Aldrich, St Louis, MO, USA). Extraction solution (25 μl) was added to each EMF tip that was incubated at 95 °C for 10 min, after which 25 μl of dilution solution was added. The ITS1f and ITS4 primers (White et al., 1990; Gardes and Bruns, 1993) were used to amplify the fungal ITS1, 5.8S and ITS2 region. Each 50 μl reaction contained 1.5 μl of template DNA, 2.5 μl of each 10 μM primer and 25 μl GoTaq master mix (Promega, Madison, WI, USA). Following an initial denaturation step of 95 °C for 3 min, amplification proceeded for 35 cycles of 94, 50 and 72 °C per min, with a final extension step of 10 min at 72 °C. PCR products were visualized on 1.4% agarose gels (SYBR Safe; Invitrogen, Carlsbad, CA, USA). PCR products producing single bands were cleaned using the Mag-Bind E-Z pure (Omega Bio-tek Inc., Norcross, GA, USA) system and sequenced (Applied Biosystems; 3130xl Genetic Analyzer, Foster City, CA, USA). If more than one band was produced (that is, from some tips morphologically identified as Cenococcum spp.), bands were extracted from the gel using an E.Z.N.A gel extraction kit (Omega Bio-tek Inc.) before cleanup and sequencing. Sequences were submitted for comparison with the GenBank databases using the BLAST algorithm (Altschul et al., 1997).
Sequencing of fungi in hyphal trap bag and FH substrates
Total community DNA was extracted from 0.5 g of all +SB, −SB and FH samples using the UltraClean Soil DNA Isolation Kit (Mo Bio Laboratories Inc., Carlsbad, CA, USA). DNA was extracted from 5 g sand samples using a previously published bead-beating method (Phillips et al., 2006) suitable for large samples. Duplicate DNA extractions were performed for each sample and pooled.
The ITS1 and partial 5.8S regions were amplified using the ITS1f (Gardes and Bruns, 1993) and ITS2r (White et al., 1990) primer pair (Integrated DNA Technologies, Coralville, IA, USA). Amplicons were generated for 454-pyrosequencing in a single-step reaction using the following fusion primers: 5′-TitaniumA-Multiplex Identifier-ITS1f-3′ and 5′-TitaniumB-ITS2r-3′ (Supplementary Table S2). Triplicate amplifications were performed for each sample. Each 50 μl amplification reaction contained 0.2 μM each primer, 200 μM dNTP mix, 2 mM MgSO4, 1 × PCR reaction buffer, 1 U Platinum Taq DNA Polymerase (Invitrogen) and 5, 10 or 20 ng of sample DNA. Following an initial denaturation step of 94 °C for 2 min, amplification proceeded for 35 cycles of 94 and 55 °C per 30 s, and 68 °C per min, with a final extension of 68 °C for 10 min. PCR products were visualized on 1.4% agarose gels (SYBR Safe; Invitrogen), replicate reactions were pooled and PCR amplicons were cleaned using the Mag-Bind E–Z pure (Omega Bio-tek Inc.) purification system.
Amplicon concentration was determined spectrophotometrically (NanoDrop ND-1000 Spectrophotometer) and both concentration and purity (that is, complete removal of primer-dimers) were verified on a 1.4% agarose gel (SYBR Safe; Invitrogen). Equimolar PCR amplicons from each sample (equivalent to 200 ng each) were pooled to create two Amplicon Libraries that which were sequenced on half a plate each using the 454 GS FLX Titanium platform (Roche Diagnostics, Basel, Switzerland) at the Génome Québec Innovation Center, Montreal, Quebec, Canada. Results were delivered as tag-sorted sequences, quality controlled for the presence of the complete primer-tag.
Bioinformatic analysis
Primer sequences were removed and additional quality filtering and trimming was performed using MOTHUR (Schloss et al., 2009). Sequences with any ambiguous (N) bases, more than eight homopolymers, shorter than 180 or longer than 400 bp, or with greater than 1 bp difference in the primer sequence were removed. Subsets of the excluded sequences were manually assessed for fungal group-specific biases. The quality filtered and trimmed sequences were then processed using the FungalITS extractor software (Nilsson et al., 2010), which extracts the variable ITS1 of the nuclear ITS1 region from the sequence, thus eliminating false alignments and clusters being generated based on the conserved 18S and 5.8S regions. The extracted ITS1 sequences were clustered at 98% (Ryberg et al., 2008) similarity (global alignment) using CD-Hit-Est (Huang et al., 2010). Representative sequences from all clusters (operational taxonomic unit, OTU) were used for BLAST searches against fully identified entries in INSD (Benson et al., 2011) and UNITE (Abarenkov et al., 2010a), using the PlutoF online server (Abarenkov et al., 2010b). A minimum of 97% similarity across 97% of the sequence length was required for positive taxonomic identification at any level. Singleton OTUs with 100% similarity to reference sequences not otherwise identified in the data set were retained, and all other singletons were discarded before statistical analysis. All sequences, fastq formatted on the Galaxy online server (Blankenberg et al., 2010), were deposited in the NCBI Short Read Archive under the accession number SRP022584.
All OTUs greater than four reads within the data set were placed into designated ecological function groups, determined by cross referencing Lawry and Diederich (2003), Rinaldi et al. (2008), Hibbet et al. (2000) and Moore et al. (2011). The groups were EMF, other mycorrhizal (ericoid, arbutoid, monotropoid and arbuscular) fungi, saprotrophs, pathogens, endophytes, yeasts and lichen fungi. Fungi known to belong to more than one of these groups were designated as mixed functional groups, whereas those whose ecological function is unverified were designated as functional group Unknown.
Statistical analysis
Before statistical analysis, 454 taxonomic data were normalized by dividing the absolute abundance of individual OTUs by total OTU’s within a sample (Gihring et al., 2012). Data were log transformed wherever required to meet the assumptions of different statistical tests (Kenkel, 2006). Relationships between the +SB, −SB, FH and sand substrates were assessed using the statistical software package PAST (v 2.17; Hammer et al., 2001). Differences in taxonomy and enzyme activity were examined by analysis of variance (Welch’s analysis of variance, if variances are unequal, as assessed using the Levene statistic), followed by a Tukey or Mann–Whitney test to determine where significant differences occurred. Relationships between enzyme activities were assessed using Spearman's r correlation test.
Cluster analysis of functional and taxonomic fungal groups was performed using a correlation matrix based on Pearson’s r correlation (1-r; PAST 2.17; Bolhuis and Stal, 2011). Species richness (S), evenness (H/ln(S)) and diversity (Fisher’s Alpha (α), Shannon (H) and Simpson’s indices) of the fungal communities were calculated using procedures available in PAST (v 2.17; Hammer et al., 2001). Patterns of enzyme activity were assessed by principle component analysis (PCA; Kenkel, 2006) in PC-ORD (McCune and Mefford, 2011). Relationships between enzyme activity and community structure were assessed by regressing enzyme PCA-loading factors with fungal community abundance (log transformed). Fungal communities significantly (P<0.05) associated with patterns of enzyme activity were illustrated as joint plot vectors overlaid on the enzyme ordinations (Ramette, 2007).
Results
General sequencing statistics
A total of 1 151 401 sequences were generated by pyrosequencing the ITS1 region of DNA extracted from hyphal trap substrates, barrier sand and forest floor samples. Quality filtering removed 362 946 sequences, primarily due to length constraints or lack of a complete primer sequence, leaving a total of 788 455 ITS1 region sequences for analysis. The 36 individually assessed samples generated an average of 32 000 sequences and an average of 22 000 quality filtered sequences (Supplementary Table S3). Clustering at 98% similarity resulted in an average of 546 OTUs per sample (singletons included), with rarefaction curves for most samples beginning to plateau (Supplementary Figure S2). The use of a stringent 98% clustering level meant that some similar OTUs were split into different clusters. These clusters were combined if they were identified through BLAST analysis, representing the same fungal OTU. A total of 5541 singletons clusters (clusters containing 1–2 sequences) that did not match any database sequences were removed from the data set (Supplementary Table S3).
Fungal taxonomic diversity
Approximately 94% of the sequences were matched to fungal sequences in the INSD and UNITE databases (Figure 1a). A total of 353 fungal OTUs were identified, including 243 fungi identified to the genus level (Supplementary Table S4). A large percentage of the identified fungi, found in all systems, were members of Basiodiomycota and Ascomycota (Figure 1a). In hyphal traps without a sand barrier (−SB) however, 44% of the sequences belonged to Fungi Incertae sedis. Hyphal traps with a sand barrier (+SB) and the surrounding FH layer contained numerous sequences that matched those of unidentified EMF amplified from root tips (21% and 11%, respectively). At this level of taxonomy, FH and +SB fungal communities clustered at a high level of similarity, whereas −SB communities formed a distinct cluster (Figure 1b). This separation was primarily due to significantly higher numbers of Fungi I. sedis in the absence of a sand barrier (Figure 1c). The presence of these fungi separated these −SB communities into distinct clusters at all levels of taxonomy (Supplementary Figure S3).

Fungal taxonomic diversity in the different hyphal trap bags, sand barriers and FH layers of the forest floor. −SB, hyphal trap bags with no sand barrier; FH, in situ fermentation-humic layer; +SB, hyphal trap bags with sand barrier; Sand, sand barrier; n=9 each. (a) The relative abundance of different phyla or group in each compartment. The total number of contributing sequence reads is indicated next to each histogram. (b) Cluster analysis of taxonomic diversity indicating the percent similarity (based on Pearson’s r correlation) of communities in the different compartments. (c) Differences in the abundance of each taxonomic group. Communities in the same row with different letters are different at the indicated significance level; NS, not significant. Unknown EMF indicates that the best matching sequences belonged to unidentified ectomycorrhizal tips. Unknown fungi indicates that the best matching sequence belonged to the kingdom Fungi.
The lowest richness and diversity occurred in −SB communities, which had significantly fewer fungal taxa than the FH samples (Table 1). The FH communities had the highest level of diversity; however, all communities were populated by a few relatively abundant taxa (Table 2). Notably, the Fungi I. sedis saprotroph Mortierella was the single-most abundant taxon (44% of sequences) found in −SB communities. By contrast, Mortierella spp. accounted for only 8% of sequences in FH and +SB communities and 16% of sequences in sand communities. Instead, these communities were largely dominated by EMF, including Piloderma spp. (FH, 32% of sequences) and Amphinema spp. (+SB, 34% of sequences; and sand, 28% of sequences). Although Piloderma spp. were the dominant EMF found in the in situ FH layer, Amphinema spp. and Wilcoxina spp. were better able to colonize both types of hyphal traps.
Fungal functional diversity
The presence of a sand barrier had a significant impact on fungal groups colonizing the hyphal traps. Hyphal traps with a sand barrier were primarily colonized by EMF, whereas hyphal traps without a sand barrier were equally colonized by saprotrophs and EMF (Figure 2a). As previously noted, −SB traps were heavily colonized by Mortierella spp., which comprised the majority of the identified saprotrophs. The trophic groups that colonized the +SB traps were highly comparable to those in the surrounding FH layer (Figure 2b), with the two groups clustering at ∼98% similarity. Both of these communities contained significantly lower populations of known saprotrophs than the −SB community (Figure 2c).

Fungal functional group diversity in the different hyphal trap bags, sand barriers and FH layers of the forest floor. −SB, hyphal trap bags with no sand barrier; FH, in situ fermentation-humic layer; +SB, hyphal trap bags with sand barrier; Sand, sand barrier; n=9 each. (a) The relative abundance of the different ecological functional groups in each compartment. The total number of contributing sequence reads is indicated next to each histogram. (b) Cluster analysis of functional group diversity, indicating the percent similarity (based on Pearson’s r correlation) of communities in the different compartments. (c) Differences in the abundance of each functional group. Communities in the same row with different letters are different at the indicated significance level; NS, not significant. Other mycorrhizae: ericoid, arbutoid, monitropoid and arbuscular mycorrhizae; mixed functional groups: belong to more than one functional group. Unknown fungi indicates that the best matching sequence belonged to the kingdom Fungi.
Ectomycorrhizal tip diversity
A total of 226 EMF tips were selected, based on abundant morphotypes, from the FH layer adjacent to the hyphal trap bags. Sequencing of the complete ITS region confirmed EMF tip identity at the following relative abundances: Cenococcum (24%), Piloderma (24%), Amphinema (17%), Tylospora (12%), Cortinarius (12%), Hygrophorus (5%) and Hysterangium (3%) spp. Approximately half of the Cenococcum tips exhibited multiple banding patterns after PCR amplification of the ITS region. Band-specific sequencing confirmed that these tips contained Cenococcum sequences in association with Phialocephala, Piloderma, Mycena or Cadophora spp. sequences (Supplementary Figure S4). For the purposes of enzyme activity assays (below), these tips were considered as Cenococcum based on the distinctive mantle formed by this fungus.
Activity of enzymes associated with SOM breakdown
The hydrolytic and oxidative enzyme activity potentials were assessed for all components of the hyphal trap systems, the adjacent FH layer and residual-activity controls (Table 3), and for EMF tips sampled from the FH layer (Table 4). Residual enzyme activity was detected for most enzymes in the gamma-irradiated control soils, which were kept under dry and sterile conditions in the lab. This residual activity was generally lower than that in the field samples, however, β-1,4-xylosidase and cellobiohydrolase activities were not statistically different from the hyphal traps (Table 3). Exploratory PCA analysis showed that activity patterns in the control samples were distinct from those in the field samples (Supplementary Figure S5), indicating that active fungi had a dominant influence on enzyme activity under the study conditions. These differences in magnitude and pattern, combined with the likelihood that residual enzymes would have been degraded under non-sterile field conditions (Allison, 2006), informed the decision not to subtract the control from field enzyme activities.
Enzyme activity in the sand barrier surrounding the +SB traps was either at or below the limit of detection, even though all sand contained visible hypha and diverse fungal communities (see Table 1). The +SB community (predominantly mycorrhizal) and the −SB community (saprotrophic and mycorrhizal) enzyme activity levels were in the same range as those of the FH community (Table 3). EMF-dominated +SB communities had significantly (P<0.01) higher levels of phenol oxidase (L-DOPA assay) and aminopeptidase activity than −SB or FH communities, and higher peroxidase activity and β-1,4-glucosidase than −SB communities. Activities of all other enzymes did not differ among fungal communities.
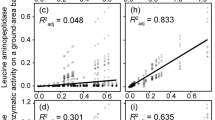
Enzyme activities associated with EMF tips were highly variable within a given genus, resulting in few significant differences between genera (Table 4). Nevertheless, Amphinema tips had significantly higher (P<0.05) enzyme activities associated with cellulose and hemicellulose degradation than did Piloderma or Cenococcum tips. The hydrolytic enzyme activity of these EMF tips was highly correlated with that of the FH layer from which they were sampled (Table 5). Both the average tip activity and the individually assessed activities of Amphinema and Piloderma spp. were positively associated with cellobiohydrolase, glucosidase and xylosidase activities within the surrounding FH layer.
Impact of fungal community structure on enzyme activity
The relationships between fungal community structure and the activity of enzymes associated with organic matter breakdown were explored by PCA. Sand samples were not included as they exhibited little enzyme activity. Differences in enzyme activity patterns between the in situ FH layer and hyphal traps were best described by the first three PCA axes (Figure 3). Location along PCA axis 1 (PC1) was driven primarily by the activities of cellulose- and hemicellulose-degrading enzymes, which strongly overlapped in the FH layer and +SB traps. Activities in these communities were also strongly associated along PC3, reflecting similar patterns of lignin degradation (peroxidase) potential. The combined effects of the PC1- and PC3-loading factors resulted in a distinct separation of the −SB traps. Both FH and −SB differed from +SB along PC2, however, reflecting variation in enzymes associated with phosphorus release, lignin degradation (laccase versus phenol oxidase) and nitrogen release.

PCA ordination of potential enzyme activity in the hyphal trap bags and the FH layers of the forest floor. The proportion of variance explained by each axis is shown, and the contribution of each enzyme in the ordination is indicated by the plot vectors. −SB, hyphal trap bags with no sand barrier; FH, in situ fermentation-humic layer; +SB, hyphal trap bags with sand barrier. Cel, cellobiohydrolase; Glu, β-1,4-glucosidase; N-Ag, β-1,4-N-acetylglucosaminidase; Pho, acid phosphatase; Sul, sulfatase; peptidase, leucine aminopeptidase; Lac, laccase; perox, peroxidase; phenox, phenol oxidase PC1 is associated with Glu (r=0.895), Cel (r=0.599), Xyl (r=0.851) and N-Ag (r=0.865); PC2 is associated with Pho (r=-0.799), peptidase (r=0.650), Lac (r=-0.695) and Phenox (r=0.729); PC3 is associated with Sul (r=0.798), peptidase (r=0.484) and Perox (r=0.702). All associations are significant at P<0.05.
The relative abundance of fungi in a given community was significantly associated (P<0.05) with patterns of enzyme activity. Joint plot vectors in Figure 4 illustrate the strength and direction of significant correlations between fungal communities and enzyme-loading factors at three different levels of community structure. At the ecological function level, the abundance of saprotrophic fungi was strongly related to the patterns of enzyme activity that separated −SB communities from the other two communities along PC1 and PC3 (Figure 4a). This association continued at the higher (Figure 4b, that is, Fungi I. sedis) and lower taxonomic levels (Figure 4c, that is, Mortierella spp.). By contrast, the enzyme-loading factors along axes PC1 and PC3 that influenced +SB and FH community clustering were generally correlated with the relative abundance of EMF and other mycorrhizal fungi. The abundance of specific EMF genera also strongly correlated with the enzyme-loading factors of PC2 (Figure 4c), which as mentioned above, largely reflects variations in enzymes associated with phosphorus and nitrogen release and lignin degradation. These fungi included Amphinema, which was strongly associated with +SB enzyme patterns, and Piloderma, Cortinarius and Tylospora, which were strongly associated with FH enzyme patterns.

Relationship between fungal communities and the potential enzyme activities in hyphal trap bags and the FH layers of the forest floor. −SB, hyphal trap bags with no sand barrier; FH, in situ fermentation-humic layer; +SB, hyphal trap bags with sand barrier. Joint plots vectors indicate the strength and direction of significant (P<0.05) correlations between enzyme PC-loading factors and fungal communities at the level of (a) ecological function, (b) phyla and (c) genus or closest identity. The direction and strength of enzyme correlations contributing to each PC axis (as in Figure 3) are indicated with the abbreviations Cel, cellobiohydrolase; Glu, β-1,4-glucosidase; N-Ag, β-1,4-N-acetylglucosaminidase; Pho, acid phosphatase; Sul, sulfatase; peptidase, leucine aminopeptidase; Lac, laccase; Perox, peroxidase.
Discussion
A large fraction of terrestrial ecosystem C pools, with estimates as high as 1700 PgC (De Luca and Boisvenue, 2012), are sequestered as SOM in the soils of temperate and boreal forests (Schmidt et al., 2011). Increased temperature and atmospheric CO2 levels associated with climate change are predicted to increase the decomposition of this SOM-C pool (Fierer et al., 2005; Conant et al., 2008; Craine et al., 2010). The majority of this SOM is found in the FH horizons dominated by EMF and not saprotrophic fungi (Lindahl et al., 2007). Quantifying the contribution of EMF mycelia to SOM turnover is, therefore, of critical importance to Cmodels (Cairney, 2012).
Potential contribution of EMF mycelia to soil organic C turnover
It has generally been believed that saprotrophic fungi bear primary responsibility for recalcitrant SOM-C decomposition in forest soils (Baldrian, 2008). Recently, Talbot et al. (2013) concluded that EMF and saprotrophic communities have complementary roles in SOM turnover. Specifically, in pine forests in a Mediterranean climate, they found that EMF species richness explained SOM-nitrogen (N) decomposition patterns, whereas saprotrophic community structure explained SOM-C decomposition patterns. In the current study, we directly measured the potential enzyme activity in FH-filled mesh bags colonized by the two different trophic groups (colonization by obligate saprotrophs controlled by the presence or absence of a sand barrier) in a sub-boreal spruce system, and compared those activities with that of the surrounding FH horizon. We found that communities dominated by EMF hyphae exhibited significant potential to degrade both SOM-C and SOM-N. The oxidative and hydrolytic enzyme activities exhibited by these EMF communities were comparable to or higher than those measured in communities enriched with saprotrophic fungi. None of the measured enzymes were specifically associated with saprotrophs, suggesting that it is premature to generalize about the role of these two trophic groups across ecosystems.
The EMF-dominated mesh bag communities and FH horizon had significantly similar patterns of cellulolytic and hemicellulolytic enzyme activity. Although these C-targeting hydrolase activities are generally attributed to saprotrophic fungi (Talbot et al., 2013), this was not the case in our spruce soils where communities with abundant saprotrophs exhibited lower hydrolytic enzyme activity. The single-most abundant taxonomic group that correlated with these activities matched unidentified EMF from root tips. Some EMF, such as Laccaria bicolor, have retained low levels of cellulolytic and hemicellulolytic hydrolases in their genomes (Martin et al., 2008; Nagendran et al., 2009; Martin et al., 2010). Although gene presence does not automatically indicate functionality, several EMF, including L. bicolor, are able to use cellulose or hemicellulose as a sole C source (Durall et al., 1994; Vaario et al., 2012). The abundant unidentified EMF in our systems may have similar hydrolytic activity, an occurrence that may be favoured by a number of site-specific factors.
Hydrolase activity is known to increase with SOM content (Sinsabaugh et al., 2008). The FH horizon in our spruce soils contained 64% SOM, almost half of which was cellulose and hemicellulose (data not shown). However, the most abundant saprotroph in our systems, Mortierella spp., is an inefficient decomposer of cellulose, exhibiting very little β-glucosidase activity (Hanson et al., 2008; Allison et al., 2009). The significantly lower β-glucosidase activity in our saprotroph-dominated hyphal traps is likely related to the abundant (50% of the total sequences) Mortierella spp. colonizing these traps. In our spruce soils, a population of inefficient saprotrophs combined with a large pool of cellulolytic compounds may have favoured hydrolytic C-prospecting by EMF, which are also known to increase their enzymatic activity in the absence of direct saprotrophic competitors (Leake et al., 2001; Pereira et al., 2012). Although we cannot exclude potential bacterial activity, a metaproteomic study of Fagus sylvatica L. litter (Schneider et al., 2012) and a 13C-cellulose stable isotope study of spruce soils (Štursová et al., 2012), both found that fungi were primarily responsible for cellulose decomposition. In the latter study, Amphinema spp. were identified as having incorporated 13C from cellulose into their DNA. In our study, the cellulose and hemicellulose-degrading activities of EMF tips, and in particular Amphinema tips, correlated highly with those activities in the FH layer. Although requiring further research, the strength of the correlations (r>0.7) and the interrelatedness of the enzymes involved make it unlikely that this is a spurious phenomenon.
Our results also suggest that EMF in spruce soils are directly involved in the breakdown of lignin and humic complexes, supporting research that suggests that increased mycorrhizal activity associated with climate change will stimulate decomposition of previously highly recalcitrant SOM-C (Cheng et al., 2012; Phillips et al., 2012b).The oxidative enzymes involved in the decomposition of these compounds are broadly classified as phenol oxidases (including laccase and tyrosinase) and peroxidases (including lignin and broad spectrum peroxidases) (Sinsabaugh, 2010; Theuerl and Buscot, 2010). Enzyme assays typically do not differentiate between the individual enzymes and instead measure the activity of the broad classes (Sinsabaugh, 2010; German et al., 2011; Eichlerová et al., 2012). In the current study, the potential phenol oxidase activity in EMF-dominated mesh bags was significantly higher than that of the FH or saprotroph-dominated bags when measured using a L-DOPA assay, but lower when measured using an ABTS assay. Research suggests that L-DOPA is oxidized by both laccases and tyrosinases, whereas ABTS is primarily oxidized by laccases (Burke and Cairney, 2002; Günther et al., 1998; Eichlerová et al., 2012). The EMF in our mesh bags did not have the relevant laccases to effectively oxidize ABTS, a fundamental difference that separated these communities from the otherwise trophically similar communities in the surrounding FH layer. These results suggest that the extent of EMF degradation of recalcitrant SOM-C will depend on the EMF community structure, as differing EMF genera were dominant in the two systems (Amphinema vs Piloderma, respectively) and individual EMF are known to differ in their ability to oxidize these enzyme substrates (Günther et al., 1998; Burke and Cairney, 2002; Tedersoo et al., 2012). Amphinema were also the most abundant EMF in the saprotroph-dominated bags, but they were outnumbered by lignin-degrading saprotrophic Mortierella spp. (Hanson et al., 2008; Allison et al., 2009). Although Allison et al. (2009) found no link between lignin degradation by Mortierella spp. and L-DOPA-phenol oxidase activity, our Mortierella-dominated communities exhibited substantial ABTS-phenol oxidase activity. Saprotrophs and EMF are known to contain a broad range of phenol oxidases that differ in substrate specificity (Luis et al., 2004; Kellner et al., 2010). Until this complex group of enzymes are better understood, researchers investigating the relative contributions of EMF and saprotrophic fungi to SOM-C turnover should include numerous phenol oxidase assays, as reviewed by Sinsabaugh (2010) in parallel.
Release of SOM-degrading enzymes by EMF does not necessarily imply the uptake of any released C. For example, phenol oxidase activity by EMF hyphae is associated with the exploration and the concomitant mining of polyphenolic-rich humic compounds for other nutrients (Talbot et al., 2008, 2013; Rineau et al., 2012). The EMF mycelia colonizing the mesh substrates may have been similarly prospecting for nitrogen, as there was a positive relationship between L-DOPA-phenol oxidase activity and protease activity (r=0.731). However, the significantly higher levels of peroxidase activity in the EMF-dominated systems were not directly associated with protease activity, suggesting that additional polyphenolic degradation may be occurring independently of nitrogen acquisition. High peroxidase activity in systems with abundant EMF is not surprising, as many EMF contain Class II peroxidase-encoding genes (Bödeker et al., 2009), and peroxidase activity has been shown to increase with increasing EMF species richness (Talbot et al., 2013). The combined and complex oxidase enzyme activity in our EMF-dominated systems suggests that EMF mycelia are actively breaking down recalcitrant SOM.
Effectiveness of modified hyphal traps to isolate EMF mycelia
Hyphal trap bags are a crucial tool for assessing the relative contributions of EMF and saprotrophic mycelium to SOM cycling (Wallander et al., 2001). Although it has been suggested that soil-based in-growth cores were primarily selected for EMF hyphae in coniferous forests with low saprotrophic populations (Hendricks et al., 2006), comparable approaches in our study (that is, bags without a sand barrier) were equally colonized by both trophic groups. Surrounding these mesh bags with a sand barrier, however, trapped a fungal community that was highly similar to that of the adjacent soil, with ∼70% EMF sequences and fewer than 9% saprotrophic sequences. Although other studies have used sand-only hyphal traps to minimize saprotroph colonization (Kjøller, 2006; Korkama et al., 2007; Wallander et al., 2010; Walker et al., 2012), in our study the sand surrounding the hyphal traps contained large populations of saprotrophic Mortierella spp. (16% of the total sequences). Mortierella were also the most abundant saprotroph in the surrounding soil and are similarly abundant in other spruce forest soils (Buée et al., 2009). These fungi are known to colonize the surface of spruce roots (Salt, 1977) and may have incidentally explored the sand in association with EMF hyphae. Our results suggest that sand-only hyphal traps may not effectively isolate EMF hyphae in soils where Mortierella are abundant.
The fungal communities in our saprotroph-excluding mesh bags were also taxonomically similar to those found in the surrounding soil, although differences did emerge at the genus level. The most abundant EMF in the hyphal traps were Amphinema and Wilcoxina, but in the surrounding soil were Piloderma, Cortinarius and Tylospora. These different colonization patterns could be because of explorer type, as EMF colonizing the +SB hyphal traps would need to explore at least the width of the sand barrier away from a colonized root tip. Although Amphinema and Piloderma are both considered medium distance explorers, Piloderma may also form mats with a limited range of exploration (Agerer, 2001; Kluber et al., 2011). Berner et al. (2012) also hypothesized that short-term mesh bag studies may preferentially select for early EMF colonizers. The most abundant EMF in our mesh bags, Amphinema and Wilcoxina, are highly competitive early colonizers of seedlings, both in greenhouses (Jones et al., 1997; Menkis et al., 2005) and under field conditions (Gagné et al., 2006; Barker et al., 2012; Walker et al., 2012). Early colonizing Amphinema and Wilcoxina may have out-competed other species, including less competitive Piloderma spp. (Erland and Söderström, 1991; Erland and Finlay, 1992; Wu et al., 1999). Differences in fungal nutritional requirements might also influence colonization. For example, Cortinarius spp. avoid nutrient-limited sand hyphal traps (Kjøller, 2006; Berner et al., 2012). These EMF may have similarly avoided the sand barrier in the current study, as Cortinarius were more abundant in hyphal traps without a sand barrier.
Conclusions
Using novel microcosms of sterile, FH substrate surrounded by a barrier of sand, we determined that both oxidative and hydrolytic enzyme activities in EMF-dominated substrates are comparable to or greater than that exhibited by microcosms enriched in saprotrophic hyphae. Collectively, our data supports a substantial role for EMF hyphae in the turnover of both recalcitrant and labile SOM in sub-boreal spruce stands. Furthermore, it expands our ability to predict feedbacks between elevated atmospheric CO2 and soil C fluxes.
Accession codes
References
Abarenkov K, Nilsson RH, Larsson K-H, Alexander IJ, Eberhardt U, Erland S et al (2010a). The UNITE database for molecular identification of fungi - recent updates and future perspectives. New Phytol 186: 281–285.
Abarenkov K, Tedersoo L, Nilsson RH, Vellak K, Saar I, Veldre V et al (2010b). PlutoF - a web based workbench for ecological and taxonomic research, with an online implementation for fungal ITS sequences. Evol Bioinform 6: 189–196.
Agerer R . (2001). Exploration types of ectomycorrhizae. Mycorrhiza 11: 107–114.
Agerer R, Rambold G . (2008) DEEMY—An information system for determination and characterisation of ectomycorrhizaewww.deemy.de.
Allison SD . (2006). Soil minerals and humic acids alter enzyme stability: implications for ecosystem processes. Biogeochemistry 81: 361–373.
Allison SD, LeBauer DS, Ofrecio MR, Reyes R, Ta A-M, Tran TM . (2009). Low levels of nitrogen addition stimulate decomposition by boreal forest fungi. Soil Biol Biochem 41: 293–302.
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W et al (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402.
Bardgett RD . (2011). Plant-soil interactions in a changing world. F1000 Biol Rep 3: 16. http://f1000.com/reports/b/3/16.
Baldrian P . (2008). Enzymes of saprotrophic basidiomycetes. In: Boddy L, Frankland JC, van West P (eds) Ecology of Saprotrophic Basidiomycetes. Academic Press: NY: USA, pp 19–41.
Barker JS, Simard SW, Jones MD, Durall DM . (2012). Ectomycorrhizal fungal community assembly on regenerating Douglas-fir after wildfire and clearcut harvesting. Oecologia 172: 1179–1189.
Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW . (2011). GenBank. Nucleic Acids Res 39: D32–D37.
Berner C, Johansson T, Wallander H . (2012). Long-term effect of apatite on ectomycorrhizal growth and community structure. Mycorrhiza 22: 615–621.
Blankenberg D, Gordon A, Von Kuster G, Coraor N, Taylor J, Nekrutenko A . (2010). Manipulation of FASTQ data with Galaxy. Bioinformatics 26: 1783–1785.
Bödeker ITM, Nygren CMR, Taylor AFS, Olson Å, Lindahl BD . (2009). ClassII peroxidase-encoding genes are present in a phylogenetically wide range of ectomycorrhizal fungi. ISME J 3: 1387–1395.
Bolhuis H, Stal LJ . (2011). Analysis of bacterial and archaeal diversity in coastal microbial mats using massive parallel 16S rRNA gene tag sequencing. ISME J 5: 1701–1712.
Brockley RP, Simpson DG . (2004). Effects of intensive fertilization on the foliar nutrition and growth of young lodgepole pine and spruce forests in the interior of British Columbia (E.P. 886.13): Establishment and progress report. B.C. Ministry of Forests, Victoria. Technical Report 018. Available at http://www.for.gov.bc.ca/hre/standman/maxprod/index.htm.
Buée M, Reich M, Murat C, Morin E, Nilsson RH, Uroz S et al (2009). 454 Pyrosequencing analyses of forest soils reveal an unexpectedly high fungal diversity. New Phytol 184: 449–456.
Burke RM, Cairney JWG . (2002). Laccases and other polyphenol oxidases in ecto- and ericoid mycorrhizal fungi. Mycorrhiza 12: 249–255.
Cairney JWG . (2012). Extramatrical mycelia of ectomycorrhizal fungi as moderators of carbon dynamics in forest soil. Soil Biol Biochem 47: 198–208.
Cheng L, Booker FL, Tu C, Burkey KO, Zhou L, Shew HD et al (2012). Arbuscular mycorrhizal fungi increase organic carbon decomposition under elevated CO2. Science 31: 1084–1087.
Clemmensen KE, Bahr A, Ovaskainen O, Dahlberg A, Ekblad A, Wallander H et al (2013). Roots and associated fungi drive long-term carbon sequestration in boreal forest. Science 339: 1615–1618.
Conant RT, Steinweg JM, Haddix ML, Paul EA, Plante AF, Six J . (2008). Experimental woaming shows that decomposition temperature sensitivity increases with soil organic matter recalcitrance. Ecology 89: 2384–2391.
Courty PE, Buée M, Diedhiou AG, Frey-Klett P, Le Tacon F, Rineau F et al (2010). The role of ectomycorrhizal communities in forest ecosystem processes. Soil Biol Biochem 42: 679–698.
Courty PE, Pritsch K, Schloter M, Hartmann A, Garbaye J . (2005). Activity profiling of ectomycorrhiza communities in two forest soils using multiple enzymatic tests. New Phytol 167: 309–319.
Craine JM, Fierer N, McLauchlan KK . (2010). Widespread coupling between the rate and temperature sensitivity of organic matter decay. Nature Geosci 3: 854–857.
De Luca TH, Boisvenue C . (2012). Boreal forest soil carbon: Distribution, function, and modelling. Forestry 85: 161–184.
Drigo B, Pijl AS, Duyts H, Kielak AM, Gamper HA, Houtekamer MJ et al (2010). Shifting carbon flow from roots into associated microbial communities in response to elevated atmospheric CO2. Proc Natl Acad Sci 107: 10938–10942.
Durall DM, Todd DM, Trappe JM . (1994). Decomposition of 14C-labelled substrates by ectomycorrhizal fungi in association with Douglas fir. New Phytol 127: 725–729.
Eichlerová I, Šnajdr J, Baldrian P . (2012). Laccase activity in soils: Considerations for the measurement of enzyme activity. Chemosphere 88: 1154–1160.
Ekblad A, Wallander H, Godbold DL, Cruz C, Johnson D, Baldrian P et al (2013). The production and turnover of extramatrical mycelium of ectomycorrhizal fungi in forest soils: role in carbon cycling. Plant Soil 366: 1–27.
Erland S, Finlay R . (1992). Effects of temperature and incubation time on the ability of three ectomycorrhizal fungi to colonize Pinus sylvestris roots. Mycol Res 96: 270–272.
Erland S, Söderström B . (1991). Effects of liming on ectomycorrhizal fungi infecting Pinus sylvestris L. III Saprophytic growth and host plant infection at different pH values in unsterile humus. New Phytol 117: 405–411.
Fierer N, Craine JM, McLauchlan KK, Schimel JP . (2005). Litter quality and the temperature sensitivity of decomposition. Ecology 86: 320–326.
Floudas D, Binder M, Riley R, Barry K, Blanchette RA, Henrissat B et al (2012). CO2 enhancement of forest productivity constrained by limited nitrogen availability. PNAS 107: 19368–19373.
Gagné A, Jany J-L, Bousquet J, Khasa DP . (2006). Ectomycorrhizal fungal communities of nursery-inoculated seedlings outplanted on clear-cut sites in northern Alberta. Can J For Res 26: 1684–1694.
Gardes M, Bruns TD . (1993). ITS primers with enhanced specificity for basidiomycetes - application to the identification of mycorrhizas and rusts. Mol Ecol 2: 113–118.
German DP, Weintraub MN, Grandy AS, Lauber CL, Rinkes ZL, Allison SD . (2011). Optimization of extracellular enzyme assay methods for ecosystem studies. Soil Biol Biochem 43: 1387–1397.
Günther Th, Perner B, Gramss G . (1998). Activities of phenol oxidizing enzymes of ectomycorrhizal fungi in axenic culture and in symbiosis with Scots pine (Pinus sylvestris L.). J Basic Microbiol 38: 197–206.
Gihring TM, Green SJ, Schadt CW . (2012). Massively parallel rRNA gene sequencing exacerbates the potential for biased community diversity comparisons due to variable library sizes. Environ Microbiol 14: 285–290.
Hagerberg D, Thelin G, Wallander H . (2003). The production of ectomycorrhizal mycelium in forests: relation between forest nutrient status and local mineral sources. Plant Soil 252: 279–290.
Hagerberg D, Wallander H . (2002). The impact of forest residue removal and wood ash amendment on the growth of the ectomycorrhizal external mycelium. FEMS Microbiol Ecol 39: 139–146.
Hammer Ø, Harper DAT, Ryan PD . (2001). PAST: Paleontological Statistics Software Packagefor Education and Data Analysis. Palaeontol Electron 4: 9.
Hanson CA, Allison SD, Bradford MA, Wallenstein MD, Treseder KK . (2008). Fungal taxa target different carbon sources in forest soil. Ecosystems 11: 1157–1167.
Hayes DJ, Turner DP, Stinson G, McGuire DA, Wei Y, West TO et al (2012). Reconciling estimates of the contemporary North American carbon balance among terrestrial biosphere models, atmospheric inversions, and a new approach for estimating net ecosystem exchange from inventory-based data. Glob Change Biol 18: 1282–1299.
Hedh J, Wallander H, Erland S . (2008). Ectomycorrhizal mycelial species composition in apatite amended and non-amended mesh bags buried in a phosphorus-poor spruce forest. Mycol Res 112: 681–688.
Hendricks JJ, Mitchell RJ, Kuehn KA, Pecot SD, Sims SE . (2006). Measuring external mycelia production of ectomycorrhizal fungi in the field: the soil matrix matters. New Phytol 171: 179–186.
Hibbet DS, Gilbert L-B, Donoghue MJ . (2000). Evolutionary instability of ectomycrorrhizal symbioses in basiomycetes. Nature 407: 506–508.
Higgins PA, Harte J . (2012). Carbon cycle uncertainty increases climate change risks and mitigation challenges. J Climate 25: 7660–7668.
Högberg MN, Högberg P . (2002). Extramatrical ectomycorrhizal mycelium contributes one-third of microbial biomass and produces, together with associated roots, half the dissolved organic carbon in a forest soil. New Phytol 154: 791–795.
Huang Y, Niu B, Gao Y, Fu L, Li W . (2010). CD-HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics 26: 680.
Intergovernmental Panel on Climate Change (2007). Climate Change 2007 Synthesis Report. Summary for Policymakers, Available at www.ipcc.ch.
Jones MD, Durall DM, Harniman SMK, Classen DC, Simard SW . (1997). Ectomycorrhizal diversity on Betula papyrifera and Pseudotsuga menziesii seedlings grown in the greenhouse or outplanted in single-species and mixed plots in southern British Columbia. Can J For Res 27: 1872–1889.
Jones MD, Phillips LA, Treu R, Ward V, Berch SM . (2012). Functional responses of ectomycorrhizal fungal communities to long-term fertilization of lodgepole pine (Pinus contorta Dougl. ex Loud. var. latifolia Engelm.) stands in central British Columbia. App Soil Ecol 60: 29–40.
Jones MD, Twieg BD, Ward V, Barker J, Durrall DM, Simard SW . (2010). Functional complementarity of Douglas-fir ectomycorrhizas for extracellular enzyme activity after wildfire or clearcut logging. Func Ecol 24: 1139–1151.
Kellner H, Zak DR, Vandenbol M . (2010). Fungi unearthed: Transcripts encoding lignocellulolytic and chitinolytic enzymes in forest soil. PLoS One 5: e10971.
Kenkel NC . (2006). On selecting an appropriate multivariate analysis. Can J Plant Sci 86: 663–676.
Kjøller R . (2006). Disproportionate abundance between ectomycorrhizal root tips and their associated mycelia. FEMS Microbiol Ecol 58: 214–224.
Kluber LA, Smith JE, Myrold DD . (2011). Distinctive fungal and bacterial communities are associated with mats formed by ectomycorrhizal fungi. Soil Biol Biochem 43: 1042–1050.
Korkama T, Fritze H, Pakkanen A, Pennanen T . (2007). Interactions between extraradical ectomycorrhizal mycelia, microbes associated with the mycelia and growth rate of Norway spruce (Picea abies) clones. New Phytol 173: 798–807.
Lawry JD, Diederich P . (2003). Lichenicolous fungi: interactions, evolution, and biodiversity. The Bryologist 106: 80–120.
Leake JR, Donnelly DP, Saunders EM, Boddy L, Read DJ . (2001). Rates and quantities of carbon flux to ectomycorrhizal mycelium following 14C pulse labelling of Pinus sylvestris L. seedlings: effects of litter patches and interaction with a wood-decomposer fungus. Tree Physio 21: 71–82.
Lensi R, Lescure C, Steinberg C, Savoie JM, Faurie G . (1991). Dynamics of residual enzyme activities, denitrification potential, and physico-chemical properties in a γ-sterilised soil. Soil Biol Biochem 23: 367–373.
Lindahl BD, Ihrmark K, Boberg J, Trumbore SE, Hogberg P, Stenlid J et al (2007). Spatial separation of litter decomposition and mycorrhizal nitrogen uptake in a boreal forest. New Phytol 173: 611–620.
Luis P, Walther G, Kellner H, Martin F, Buscot F . (2004). Diversity of laccase genes from Basidiomycetes in a forest soil. Soil Biol Biochem 36: 1025–1036.
Martin F, Aerts A, Ahrén D, Brun A, Duchaussoy F, Kohler A et al (2008). The genome sequence of the basidiomycete fungus Laccaria bicolor provides insights into the mycorrhizal symbiosis. Nature 452: 88–92.
Martin F, Kohler A, Murat C, Balestrini R, Coutinho PM, Jaillon O et al (2010). Perigord black truffle genome uncovers evolutionary origins and mechanisms of symbiosis. Nature 464: 1033–1038.
McCune B, Mefford MJ . (2011) PC-ORD. Multivariate Analysis of Ecological Data. Version 6.0. MjM Software: Gleneden Beach, Oregon, USA.
Menkis A, Vasiliauskas R, Taylor AFS, Stenlid J, Finlay R . (2005). Fungal communities in mycorrhizal roots of conifer seedlings in forest nurseries under different cultivation systems, assessed by morphotyping, direct sequencing and mycelial isolation. Mycorrhiza 16: 33–41.
Moore D, Robson GD, Trinci APJ . (2011) 21st Century Guide to Fungi. Cambridge University Press: Cambridge, UK.
Myneni RB, Dong J, Tucker CJ, Kaufmann RK, Kauppi PE, Liski J et al (2001). A large carbon sink in the woody biomass of Northern forests. Proc Natl Acad Sci USA 98: 14784–14789.
Nagendran S, Hallen-Adams HE, Paper JM, Aslam N, Walton JD . (2009). Reduced genomic potential for secreted plant cell-wall-degrading enzymes in the ectomycorrhizal fungus Amanita bisporigera, based on the secretome of Trichoderma reesei. Fungal Genet Biol 46: 427–435.
Nilsson RH, Veldre V, Hartmann M, Unterseher M, Amend A, Bergsten J et al (2010). An open source software package for automated extraction of ITS1 and ITS2 from fungal ITS sequences for use in high-throughput community assays and molecular ecology. Fungal Ecol 3: 284–287.
Norby RJ, Warren JM, Iversen CM, Medlyn BE, McMurtrie RE . (2010). CO2 enhancement of forest productivity constrained by limited nitrogen availability. PNAS 107: 19368–19373.
Orwin KH, Kirschbaum MUF St, John MG, Dickie IA . (2011). Organic nutrient uptake by mycorrhizal fungi enhances ecosystem carbon storage: a model based assessment. Ecol Lett 14: 493–502.
Pereira E, Coelho V, Tavares RM, Lino-Neto T, Baptista P . (2012). Effect of competitive interactions between ectomycorrhizal and saprotrophic fungi on Castanea sativa performance. Mycorrhiza 22: 41–49.
Phillips CL, Kluber LA, Martin JP, Caldwell BA, Bond BJ . (2012a). Contributions of ectomycorrhizal fungal mats to forest soil respiration. Biogeosciences 9: 2099–2110.
Phillips LA, Greer CW, Germida JJ . (2006). Culture-based and culture-independent assessment of the impact of mixed and single plant treatments on rhizosphere microbial communities in hydrocarbon contaminated flare-pit soil. Soil Biol Biochem 38: 2823–2833.
Phillips RP, Meier IC, Bernhardt ES, Grandy AS, Wickings K, Finzi AC . (2012b). Roots and fungi accelerate carbon and nitrogen cycling in forests exposed to elevated CO2. Ecol Lett 15: 1042–1049.
Potila H, Wallander H, Sarjala T . (2009). Growth of ectomycorrhizal fungi in drained peatland forests with variable P and K availability. Plant Soil 316: 139–150.
Pritsch K, Raidl S, Marksteiner E, Agerer R, Blaschke H, Schloter M et al (2004). A rapid and highly sensitive method for measuring enzyme activities in single mycorrhizal tips using 4-methylumbelliferone-labelled fluorogenic substrates in a microplate system. J Microbiol Meth 58: 233–241.
Ramette A . (2007). Multivariate analyses in microbial ecology. FEMS Microbiol Ecol 62: 142–160.
Rinaldi AC, Comandini O, Kuyper TW . (2008). Ectomycorrhizal fungal diversity: separating the wheat from the chaff. Fungal Divers 33: 1–45.
Rineau F, Roth D, Shah F, Smits M, Johansson T, Canbäck B et al (2012). The ectomycorrhizal fungus Paxillus involutus converts organic matter in plant litter using a trimmed brown-rot mechanism involvinetg Fenton chemistry. Environ Microbiol 14: 1477–1487.
Ryberg M, Nilsson RH, Kristiansson E, Töpel M, Jacobsson S, Larsson E . (2008). Mining metadata from unidentified ITS sequences in GenBank: A case study in Inocybe (Basidiomycota). BMC Evol Biol 8: 50–64.
Saiya-Cork KR, Sinsabaugh R, Zak DR . (2002). The effects of long term nitrogen deposition on extracellular enzyme activity in an Acer saccharum forest soil. Soil Biol Biochem 34: 1309–1315.
Salt GA . (1977). The incidence of root-surface fungi on naturally regenerated Picea sitchensis seedlings in southeast Alaska. Forestry 50: 113–115.
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB et al (2009). Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75: 7537–7541.
Schmidt WI, Torn MS, Abiven S, Dittmar T, Guggenberger G, Janssens IA et al (2011). Persistence of soil organic matter as an ecosystem property. Nature 478: 49–56.
Schneider T, Keiblinger KM, Schmid E, Sterflinger-Gleixner K, Ellersdorfer G, Roschitzki B et al (2012). Who is who in litter decomposition? Metaproteomics reveals major microbial players and their biogeochemical functions. ISME J 6: 1749–1762.
Sinsabaugh RL . (2010). Phenol oxidase, peroxidase and organic matter dynamics of soil. Soil Biol Biochem 42: 391–404.
Sinsabaugh RL, Lauber CL, Weintraub MN, Ahmed B, Allison SD, Crenshaw C et al (2008). Stoichiometry of soil enzyme activity at global scale. Ecol Lett 11: 1252–1264.
Sinsabaugh RL, Saiya-Cork K, Long T, Osgood MP, Neher DA, Zak DR et al (2003). Soil microbial activity in a Liquidambar plantation unresponsive to CO2- driven increases in primary production. Appl Soil Ecol 24: 263–271.
Štursová M, Žifčaková L, Leigh MB, Burgess R, Baldrian P . (2012). Cellulose utilization in forest litter and soil: identification of bacterial and fungal decomposers. FEMS Microbiol Ecol 80: 735–746.
Talbot JM, Allison SD, Treseder KK . (2008). Decomposers in disguise: mycorrhizal fungi as regulators of soil C dynamics in ecosystems under global change. Funct Ecol 22: 955–963.
Talbot JM, Bruns TD, Smith DP, Branco S, Glassman SI, Erlandson S et al (2013). Independent roles of ectomycorrhizal and saprotrophic communities in soil organic matter decomposition. Soil Biol Biochem 57: 282–291.
Tedersoo L, Naadel T, Bahram M, Pritsch K, Buegger F, Leal M et al (2012). Enzymatic activities and stable isotope patterns of ectomycorrhizal fungi in relation to phylogeny and exploration types in an afrotropical rain forest. New Phytol 195: 832–843.
Theuerl S, Buscot F . (2010). Laccases: Toward disentangling their diversity and functions in relation to soil organic matter cycling. Biol Fertil Soils 46: 215–225.
Treseder KK, Torn MS, Masiello CA . (2006). An ecosystem-scale radiocarbon tracer to test use of litter carbon by ectomycorrhizal fungi. Soil Biol Biochem 38: 1077–1082.
Vaario L-M, Heinonsalo J, Spetz P, Pennanen T, Heinonen J, Tervahauta A et al (2012). The ectomycorrhizal fungus Tricholoma matsutake is a facultative saprotroph in vitro. Mycorrhiza 22: 409–418.
van der Wal A, Geydan TD, Kuyper TW, de Boer W . (2012). A thready affair: linking fungal diversity and community dynamics to terrestrial decomposition processes. FEMS Microbiol Rev 37: 477–494.
Von Lützow M, Kögel-Knabner I . (2009). Temperature sensitivity of soil organic matter decomposition—what do we know? Bio Fertil Soil 46: 1–15.
Walker JKM, Ward V, Paterson C, Jones MD . (2012). Coarse woody debris retention in subalpine clearcuts affects ectomycorrhizal root tip community structure within 15 years of harvest. Appl Soil Ecol 60: 5–15.
Wallander H, Johansson U, Sterkenburg E, Durling MB, Lindahl BD . (2010). Production of ectomycorrhizal mycelium peaks during canopy closure in Norway spruce forests. New Phytol 187: 1124–1134.
Wallander H, Nilsson L-O, Hagerberg D, Bååth E . (2001). Estimation of the biomass and seasonal growth of external mycelium of ectomycorrhizal fungi in the field. New Phytol 151: 753–760.
White TJ, Bruns TD, Lee S, Taylor J . (1990). Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand DH (eds) PCR Protocols: A Guide to Methods and Applications. Academic Press: London, UK, pp 315–322.
Wolfe BE, Tulloss RE, Pringle A . (2012). The irreversible loss of a decomposition pathway marks the single origin of an ectomycorrhizal symbiosis. PLoS One 7: e39597.
Wright DP, Johansson T, Le Quere A, Soderstrom B, Tunlid A . (2005). Spatial patterns of gene expression in the extramatrical mycelium and mycorrhizal root tips formed by the ectomycorrhizal fungus Paxillus involutus in association with birch (Betula pendula) seedlings in soil microcosms. New Phytol 167: 579–596.
Wu B, Nara K, Hogetsu T . (1999). Competition between ectomycorrhizal fungi colonizing Pinus densiflora. Mycorrhiza 9: 151–159.
Acknowledgements
This study was funded by a Natural Sciences and Engineering Research Council of Canada Strategic Grant. The preliminary study development, additional field and lab assistance was provided by Jeremy Bougoure, Tristyn Hay, Lindsay Reddeman and Jennifer Walker. We thank NetPak of Coquitlam, British Columbia, Australia for donating the plastic containers used to construct the hyphal traps with sand barriers. We acknowledge Alexandre Montpetit of the genotyping platform of the McGill University and Génome Québec Innovation Centre for their technical assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article
Cite this article
Phillips, L., Ward, V. & Jones, M. Ectomycorrhizal fungi contribute to soil organic matter cycling in sub-boreal forests. ISME J 8, 699–713 (2014). https://doi.org/10.1038/ismej.2013.195
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2013.195
Keywords
This article is cited by
-
The conversion of mycorrhizal types closely associated with the changes in microbial keystone taxa and potential function in subtropical forests
Plant and Soil (2023)
-
Co-invasive ectomycorrhizal fungi alter native soil fungal communities
Plant and Soil (2023)
-
Maize/peanut intercropping improves nutrient uptake of side-row maize and system microbial community diversity
BMC Microbiology (2022)
-
Global systematic review with meta-analysis shows that warming effects on terrestrial plant biomass allocation are influenced by precipitation and mycorrhizal association
Nature Communications (2022)
-
Molecular Characterization of Distinct Fungal Communities in the Soil of a Rare Earth Mining Area
Microbial Ecology (2022)
