
Abstract
Gut microbiota of invasive Asian silver carp (SVCP) and indigenous planktivorous gizzard shad (GZSD) in Mississippi river basin were compared using 16S rRNA gene pyrosequencing. Analysis of more than 440 000 quality-filtered sequences obtained from the foregut and hindgut of GZSD and SVCP revealed high microbial diversity in these samples. GZSD hindgut (GZSD_H) samples (n=23) with >7000 operational taxonomy units (OTUs) exhibited the highest alpha-diversity indices followed by SVCP foregut (n=15), GZSD foregut (n=9) and SVCP hindgut (SVCP_H) (n=24). UniFrac distance-based non-metric multidimensional scaling (NMDS) analysis showed that the microbiota of GZSD_H and SVCP_H were clearly separated into two clusters: samples in the GZSD cluster were observed to vary by sampling location and samples in the SVCP cluster by sampling date. NMDS further revealed distinct microbial community between foregut to hindgut for individual GZSD and SVCP. Cyanobacteria, Proteobacteria, Actinobacteria and Bacteroidetes were detected as the predominant phyla regardless of fish or gut type. The high abundance of Cyanobacteria observed was possibly supported by their role as the fish’s major food source. Furthermore, unique and shared OTUs and OTUs in each gut type were identified, three OTUs from the order Bacteroidales, the genus Bacillariophyta and the genus Clostridium were found significantly more abundant in GZSD_H (14.9–22.8%) than in SVCP_H (0.13–4.1%) samples. These differences were presumably caused by the differences in the type of food sources including bacteria ingested, the gut morphology and digestion, and the physiological behavior between GZSD and SVCP.
Similar content being viewed by others

Introduction
The silver carp (SVCP; Hypophthalmichthys molitrix) have been introduced as a food fish or a way to control plankton populations in many countries (Kolar, 2007; Sampson et al., 2009). In the United States, SVCP have escaped captivity, successfully invaded much of the Mississippi river basin (Chick and Pegg, 2001), and dominated these aquatic communities. It is reported that SVCP could effectively compete with native fishes such as the planktivorous gizzard shad (GZSD; Dorosoma cepedianum) for food resources in the Illinois river (Irons et al., 2007). Should SVCP populations successfully establish, populations of native fishes would decline and ultimately result in potentially irreversible ecological changes in the United States (Chen et al., 2007; Herborg et al., 2007).
Many efforts have been made to control SVCP populations in the United States (Chick and Pegg, 2001). As a part of this process, it is essential to obtain greater understanding of the indigenous and invasive fish’s behavior (Chick and Pegg, 2001). One approach for unveiling animal behavior is to examine the gut microbial community (Hooper et al., 2001; Ringų et al., 2003); many analogous studies on human (Qin et al., 2010; Wang et al., 2011), mice (Zhang et al., 2009), macaque (McKenna et al., 2008), chicken (Lan et al., 2005; Torok et al., 2008), earthworm (Drake et al., 2006) and termite (Ohkuma and Brune, 2011) successfully correlated gut microbial communities to the host’s physiology. Therefore, understanding the gut flora of SVCP and native fish would certainly provide insight into behavioral differences and perhaps solutions for controlling SVCP invasion in the United States.
So far, the microbial community in fish guts has not been systematically characterized. Most previous studies used traditional culture-dependent or microscopy methods to investigate the fish gut microbial diversity (Xu et al., 1988; Ringų et al., 2003). These conventional methods are hampered by the inability to cultivate these microorganisms (Hugenholtz et al., 1998). With the development of the next-generation sequencing technologies, large number of DNA sequences can be obtained for a targeted DNA biomarker (Metzker, 2009). Among all high-throughput sequencing platforms available, 454 pyrosequencing technology provides relatively long read length with a low error rate, and has been widely used to investigate microbial communities on the basis of the 16S rRNA gene sequences (Roesch et al., 2007; Claesson et al., 2009; Hong et al., 2010; Ye et al., 2011).
Little is known about the gut microbial community differences between American indigenous fish and invasive SVCP in the United States. So far, only one study has applied the 16S pyrosequencing approach to characterize the microbial community in the gut of grass carp in China (Wu et al., 2012). Thus, this study was set to investigate the intestinal microbial flora from invasive SVCP and GZSD (an important indigenous planktivore), obtained in the middle region of the Mississippi river basin, to better understand the microbial complexities and diversities of their gut microbiota. It is hypothesized that the fish gut flora and the fecal materials discharged into the water can reflect their diet preferences, physiological behaviors and presence in the river. Furthermore, knowing the microbial community in the fish guts of Asian carp and native fishes can provide useful information on how to monitor better and manage Asian carp populations. For example, host-specific biomarkers can be discovered and designed to determine the presence and quantity of Asian carp and native fishes in the freshwater. It is expected that the biomarker based on the gut microbial species can increase the detection sensitivity, as the amount of fecal matter from Asian carp is abundant in the waters. This method can further complement existing molecular surveillance techniques, like environmental DNA (Jerde et al., 2011).
Materials and methods
Any use of a trade, product or company name in this study is for descriptive purposes only and does not imply endorsement by the US Government.
Sample collection and DNA extraction
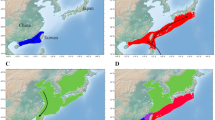
In this study, 24 GZSD and 28 SVCP were caught from Illinois river (Havana, IL, USA), Ted Shanks Conservation Area wetlands (Louisiana, MO, USA), Wabash River (West Lafayette, IN, USA) and Perche Creek (McBaine, MO, USA) in the United States in August, September and October of 2010, and in May, June, August, September and October of 2011 (Figure 1a and Supplementary Table S1). During these sampling times, average water temperatures varied from 14.0 °C in May to 17.9 °C in August. The weight and length of GZSD were from 0.28 to 0.35 kg and from 291 to 312 mm, respectively, and those of SVCP were from 1.05 to 3.20 kg and 477–678 mm, respectively. The sex ratio of both fish species was about half male and half female. Prior to dissection, all fish were euthanized with an overdose of tricaine methanesulfonate (Argent Chemical Laboratories, Redmond, WA, USA) (Hansen et al., 2006) or by cranial concussion (Amberg et al., 2012). All procedures for handling and euthanasia of animals were approved by the US Geological Survey Upper Midwest Environmental Sciences Center Animal Care and Use Committee. Foreguts (that is, portion of intestinal tract posterior to bile duct to the first distal loop) and hindguts (that is, intestinal tract anterior from anus to last anterior loop) were removed. The gut contents were carefully collected into plastic cryo-tubes, flash frozen and stored at −80 °C before they were transported to the laboratory and stored at −20 °C. The type of fish gut samples included GZSD foregut (GZSD_F), GZSD hindgut (GZSD_H), SVCP foregut (SVCP_F) and SVCP hindgut (SVCP_H). Genomic DNA of each sample was extracted using a FastDNA spin kit for soil (Qbiogene, Carlsbad, CA, USA) based on the manufacturer’s instruction. Extracted DNA was further purified with the Promega Wizard DNA clean-up system (Promega, Madison, WI, USA).

Sampling location, gut diagrams and microbial diversity. (a) Sampling locations. (b) Rarefaction curve sequences showing the microbial community complexities in the guts of GZSD at 3% distance cutoff. (c) Rarefaction curve sequences showing the microbial community complexities in the guts of SVCP at 3% distance cutoff. Fish illustrations were gifts from Duane Raver.
454 pyrosequencing and data analysis
In total, 104 genomic DNA samples were obtained from the foregut and hindgut contents of 24 GZSD and 28 SVCP. A primer set (515F/909R) targeting the 16S rRNA gene sequences was used for PCR amplification as described previously (Tamaki et al., 2011). On the basis of the quality check of DNA extraction and PCR amplification, only 71 samples could be successfully amplified with high-quality 16S rRNA gene PCR products (Supplementary Table S1 and Table 1). The amplified PCR products were separated by 1.5% low-melting gel electrophoresis and extracted with a Wizard SV Gel and PCR clean-up system (Promega, St Louis, MO, USA). A Qubit fluorometer (Invitrogen, Carlsbad, CA, USA) was used to quantify PCR products. Equal amounts of the PCR products were pooled together and sequenced on a 454 GS FLX Titanium platform (Roche Applied Science, Indianapolis, IN, USA) at the Biotechnology Center at the University of Illinois at Urbana-Champaign.
Quality filtering, denoising and chimera checking of the sequences obtained from pyrosequencing were conducted using the Quantitative Insights Into Microbial Ecology (QIIME) v 1.5.0 pipeline (Caporaso et al., 2010). The rarefaction curves and the alpha-diversity indices (that is, Chao 1 estimator, abundance-based coverage estimator and Shannon estimator) were calculated using Mothur (Schloss et al., 2009). The taxonomic assignment of the sequences were performed using RDP classifier (Wang et al., 2007) with 80% confidence level. Visualization of microbial communities in different fish gut samples was conducted using Circos (Krzywinski et al., 2009) and iTOL (Letunic and Bork, 2007). The shared and unique operational taxonomy unit (OTU) analysis was conducted on the basis of the OTU table generated by QIIME. The shared OTUs were defined as those that were present in at least a certain percentage (30 or 40%) of the samples of each kind of fish gut. For example, if an OTU was present in more than 30% of those GZSD_F and 30% of GZSD_H samples but not in SVCP_F and SVCP_H samples, it was defined as an OTU shared by GZSD_F and GZSD_H. The unique OTUs were arbitrarily defined as those that were only present in more than 30% (or 40%) of those samples taken from one type of fish gut sample and were not found in the other three type of fish gut samples. It was observed that a cutoff higher than 40% greatly reduced the number of shared and unique populations.
Unclassified 16S rRNA gene sequences at the phylum level, and the Cyanobacteria- and Bacteroidetes-related 16S rRNA gene sequences were further extracted from the entire sequences and clustered into OTUs at different homologies (97% for the unclassified sequences, and 100% for Cyanobacteria and Bacteroidetes sequences) using uclust (Edgar, 2010) method in QIIME. The representative sequences for these three categories were picked and aligned using Greengenes’s NAST server (DeSantis et al., 2006). The aligned sequences were imported into ARB and then added to the Greengenes ARB database (Greengenes_16S_2011_1.arb) using ARB parsimony method. Phylogenetic trees of the sequences in this study and their neighbors in the Greengenes ARB database were constructed using neighbor-joining method with Jukes−Cantor corrected DNA distances.
Statistical analysis
Distances between microbial communities in different samples were calculated using the weighted UniFrac beta-diversity metric via QIIME. Non-metric multidimensional scaling (NMDS) was used to visualize the pairwise UniFrac distances among samples. To identify OTUs that exhibited significant differences in abundance between different fish gut samples, Welch’s t-test (confidence interval method: Welch’s inverted, P<0.05) for two samples having possibly unequal variances (Welch, 1947) was performed within STAMP (Parks and Beiko, 2010).
Results
Microbial complexity of fish gut flora
A total of 441 601 quality-filtered sequences were obtained from 71 samples with the number of sequences ranging from 2042 to 15 749 per sample (Supplementary Table S1). After removing chimera sequences (∼10% of the total reads), the microbial complexity in the hindgut and foregut of SVCP and GZSD were estimated on the basis of alpha-diversity indices (that is, abundance-based coverage estimator, Chao1 and Shannon indices). GZSD_H samples had the largest alpha-diversity indices (abundance-based coverage estimator, 9254; Chao1, 7478; and Shannon, 6.80 based on subsampled data), followed by GZSD_F samples and then the SVCP_F samples (Table 1). SVCP_H samples had the lowest values (abundance-based coverage estimator, 3329; Chao1, 2266; and Shannon, 3.4 based on subsampled data). Similar trends in microbial diversity were observed among the four gut types based on the rarefaction curves (Figures 1b and c) and the number of known taxa assigned (Supplementary Figure S1). Greater numbers of OTU and known taxa, at different phylogenetic levels, were observed in GZSD_H, followed by GZSD_F, SVCP_F and SVCP_H.
Microbial community compositions
Figure 2 illustrates the abundance of 12 major phyla commonly observed in all four fish gut types in a circular viewer. It shows that within each fish gut type, 4.2–9.1% of total sequences could not be assigned to known microbial phyla. The phylum Cyanobacteria/Chloroplast was one of the prominent phyla, representing 33.5, 12.8, 27.4 and 27.5% of total sequences in GZSD_F, GZSD_H, SVCP_F and SVCP_H, respectively. Actinobacteria accounted for 22.8, 17.1 and 5.3% of total sequences in GZSD_F, GZSD_H and SVCP_F, respectively, but was only 0.9% in SVCP_H. The abundance of Proteobacteria ranged from 15.0 to 21.1% in GZSD_F, GZSD_H and SVCP_F, but decreased to 2.1% in SVCP_H. Other prominent phyla included Firmicutes (5.7–25.7%), Planctomycetes (0.1–2.4%), Chloroflexi (0.1–5.7%), Crenarchaeota (0–2.2%), Bacteroidetes (1.4–25.3%) and Fusobacteria (0.9–11.8%).

Circular representation of microbial communities in fish gut samples at phylum level and OTU level. The inner circular diagram shows the relative abundance of different phyla in fish gut samples. Phyla with abundance lower than 1% in all samples were not shown. Sequences that could not be assigned at phylum level were marked as ‘Unknown’. The peripheral bubble plot shows the dominant OTUs in each phylum. The bubble plot consists of four layers, which from inside to outside represent GZSD_F, GZSD_H, SVCP_F and SVCP_H. Only OTUs with relative abundances over 0.5% in at least one sample were shown here. The percentages ⩽3% were shown in dots with different sizes and the percentages >3% were indicated by numbers.
The phylogenetic affiliation of those OTUs with abundance >0.01% in Proteobacteria, Fusobacteria, Firmicutes, Actinobacteria, Cyanobacteria, Bacteroidetes and Crenarchaeota was further shown with bubble plot in Figure 2. Nine OTUs had an abundance >3% in at least one fish gut type. Among them, OTU-3807 (Bacillariophyta genus) was prominent in all fish gut types. In addition, numerous OTUs were observed to be five times more abundant in one fish gut type than the others. Six OTUs (OTU-0080, 2358, 2378, 3189, 3368 and 7018) could not be assigned to known phyla by RDP classifier. However, these six OTUs were successfully classified to Cyanobacteria, Firmicutes and Chloriflexi using the phylogeny tool ARB (Supplementary Figure S2).
Approximately 7.2% of total sequencing reads could not be classified at phylum level using RDP classifier. Using QIIME, these sequences were reclassified into 2155 OTUs at a similarity of 97%. Among them, 22 OTUs contained >100 sequences within each OTU, and the total number of sequences in the 22 OTUs accounted for 56.1% of the total unclassified sequences. Representative sequences from those 22 OTUs were used to construct a phylogeny tree (Supplementary Figure S3 and Supplementary Table S2). Among them, five OTUs were affiliated with Firmicutes (35.5% of total unclassified sequences), two with Nitrospirae (1.3%), six with Cyanobacteria (10.0%), three with Proteobacteria (3.2%), three with Chloriflexi (3.1%) and two with Plactomycetacia (0.5%). These results indicated that those fish gut samples did not contain a large number of novel or candidate microbial phyla.
Microbial community similarity between fish gut types
NMDS plot was used to compare the similarity in the microbial community compositions of hindgut samples taken from SVCP and GZSD. Among all hindgut samples, GZSD_H formed a distinct cluster and was separated from all SVCP_H samples (Figure 3). The clustering pattern among GZSD_H samples was primarily influenced by sampling location with samples from three locations forming three distinct clusters. The sampling time did not affect the clustering pattern. Although the SVCP_H samples were also collected from different locations, the clustering pattern was mostly affected by sampling time, with distinct clusters observed for samples taken from June and October. Samples taken from August and September were closer to the June samples than to the October samples.

NMDS plot showing the microbial community differences between GZSD_H and SVCP_H samples. Pairwise community distances were determined using the weighted UniFrac algorithm.
The differences in microbial community between foregut and hindgut taken from each single fish were examined (Supplementary Figure S4). Results showed no distinct pattern or clustering between GZSD_F and SVCP_F (Supplementary Figure S4), but exhibited clear microbial community shifts from foregut to hindgut (Supplementary Figure S5). The normalized weighted UniFrac distances between foregut and hindgut of a fish ranged from 0.30 to 0.80 (average: 0.55) for SVCP and from 0.28 to 0.90 (average: 0.60) for GZSD, suggesting a significant change in the microbial community composition between the foregut and the hindgut (Supplementary Table S3). The relatedness between the foregut and hindgut microflora taken from individual fish was determined by calculating the correlation of abundances of the top 20 OTUs in the foregut and hindgut of each individual fish. The correlations between the foregut and hindgut of GZSD and SVCP were very low for most fishes (R2<0.8), suggesting that the microbial community structures in the foregut and hindgut were different (Supplementary Table S3).
Shared and unique microbial populations
To investigate the microbial community in different fish gut samples, the shared and unique OTUs were analyzed through a Venn diagram. Pairwise comparison was first performed among all gut types by considering the shared OTUs, as those present in at least 30% of the samples of each gut type, and the unique OTUs as those only present in more than 30% of the samples within a gut type. The number of shared OTUs varied from 34 to 187 and unique OTUs varied from 3 to 258 (Figure 4a). By increasing the cutoff for shared and unique OTUs to 40%, the number of shared and unique OTUs decreased noticeably with the shared OTUs ranging from 20 to 114 and the unique OTUs from 0 to 125. Figure 4b shows the shared and unique OTUs of the four kind of fish gut samples at a cutoff of 30%. Only a few OTUs (ranging from 1 to 26) were uniquely present in one type of fish gut sample. All these unique OTUs had very low abundance, accounting for only 0.02–0.27% of the total sequences. In contrast, the sequences in the OTUs shared by the four kind of fish gut samples represented 47.8% of the total sequences.

Unique and shared OTUs in GZSD and SVCP gut samples. (a) The amount of shared and unique OTUs between pairwise GZSD and SVCP gut samples at 30% and 40% cutoff level. For each bar plot, the bar in the center represents the number of shared OTUs in the pairwise samples and the bar on the side represents the number of unique OTUs in the corresponding sample. (b) Venn diagram showing the number of shared and unique OTUs among all the four types of GZSD and SVCP gut samples at 30% cutoff level. The percentages in the Venn diagram indicate the ratios of the sequences that are associated to the OTUs in total sequences. The definitions of the shared and unique OTUs were present in the Materials and Methods section.
Welch’s t-test was further used to identify 18 OTUs that were significantly different in abundances in the foreguts and hindguts of GZSD and SVCP (Figure 5). Among them, seven OTUs were abundantly present in SVCP_H, and the remaining had higher abundances in GZSD_H. In particular, OTU-9252 (Bacteroidales order), OTU-3807 (Bacillariophyta genus), OTU-9374 (Clostridium genus) had significantly higher abundance in GZSD_H (14.9–22.8%) than in SVCP_H (0.13–4.1%). Supplementary Table S4 indicates that between GZSD_F and SVCP_F, 12 OTUs exhibited significant differences in abundances, but the differences (0.5–1.7%) were relatively low. Five OTUs had significant difference in abundances between GZSD_F and GZSD_H, and the largest difference was observed with OTU-0450 from the phylum Firmicutes (7.5%). Seven OTUs had significantly different abundances between SVCP_F and SVCP_H. OTU-9252 (Bacteroidales order) had significantly higher abundance in SVCP_H than in SVCP_F (16.5%).

Extended error bar plot showing the OTUs that have significantly different abundances in the hindguts of GZSD and SVCP. OTUs overrepresented in the SVCP samples have a negative difference between relative abundances and OTUs overrepresented in the GZSD samples have a positive difference between relative abundances. Only OTUs with mean proportions larger than 0.5% in the two groups of samples were shown.
Cyanobacteria and Bacteroidetes
Cyanobacteria and Bacteroidetes were two important phyla mainly responsible to the differences among all fish gut samples. Sequences affiliated with Cyanobacteria and Bacteroidetes were further retrieved from the denoised sequence data and used to identify unique OTUs related to SVCP and GZSD. Using a sequence similarity cutoff of 100%, 1860 and 1084 OTUs were affiliated with Cyanobacteria and Bacteroidetes, respectively. Among them, 28 Cyanobacteria OTUs and 27 Bacteroidetes OTUs were present only in GZSD_H samples, and 18 Cyanobacteria OTUs and 26 Bacteroidetes OTUs were present only in SVCP_H samples (OTUs with less than 10 sequences were not considered). Phylogeny trees using representative sequences of these OTUs (Supplementary Figures S6 and S7) indicate that most Bacteroidetes OTUs from GZSD and SVCP were grouped into different clusters unique to SVCP or GZSD. In contrast, GZSD and SVCP shared almost all common Cyanobacteria OTUs, except one distinct cluster formed by 11 OTUs in SVCP samples.
Discussion
Although studies on fish gut microbiota have been reported (Roeselers et al., 2011; Sullam et al., 2012; Wu et al., 2012), little is known about the gut microbial community difference between invasive Asian carp and indigenous fish. The number of OTUs observed within the foregut and hindgut of SVCP and GZSD in this study are equal or higher than those reported in macaque gut samples (McKenna et al., 2008), grass carp hindgut (Wu et al., 2012) and zebrafish (Roeselers et al., 2011) (Figure 6a). The microbial diversity observed with GZSD gut flora is also higher than those in Asian carp and human gut flora, and comparable with a soil sample collected from Illinois, USA (Roesch et al., 2007). An NMDS plot (Figure 6b) suggests that the microbial community of GZSD_H is closer to grass carp (Wu et al., 2012) and zebrafish (Roeselers et al., 2011) than to GZSD_F, and SVCP_H flora is not closely related to other gut flora. The microbial composition found in the foregut and hindgut of SVCP and GZSD was further compared with other gut flora studied so far at the phylum level. Cyanobacteria were the most dominant phylum found only in the gut of GZSD, SVCP and grass carp (Wu et al., 2012), and are mostly absent or very low in guts of terrestrial vertebrates (Supplementary Figure S8). All fish gut samples in this study and most fish gut samples in previous studies (Roeselers et al., 2011; Sullam et al., 2012; Wu et al., 2012) shared Proteobacteria and Firmicutes as the most dominant phyla. Actinobacteria were mainly found in GZSD, grass carp (Wu et al., 2012) and rainbow trout (Huber et al., 2004; Kim et al., 2007; Sullam et al., 2012). Euryarchaeota are unique to only GZSD. Bacteroidetes were abundant in SVCP, marine herbivorous fish (Sullam et al., 2012) and other non-fish gut flora (Supplementary Figure S8).

Microbial community complexity and similarity in different fish gut systems. (a) Comparison of OTU number in different gut and environmental samples on the basis of 3000 sequences at 3% distance cutoff. (1) SVCP_H, (2) SVCP_F, (3) GZSD_F, (4) GZSD_H, (a) zebrafish (Roeselers et al., 2011, b) grass carp intestinal mucosa (Wu et al., 2012, c) macaque lower gastrointestinal mucosal surface (McKenna et al., 2008, d) grass carp intestinal content (Wu et al., 2012, e) human distal intestine (Claesson et al., 2009, f) soil samples collected from Illinois, USA (Roesch et al., 2007). (b) NMDS plot showing the microbial community differences from different gut flora (grass carp (Wu et al., 2012), beef cattle (Shanks et al., 2011), broiler chicken (Danzeisen et al., 2011), human (Claesson et al., 2009), dog (Middelbos et al., 2010), swine (Looft et al., 2012) and zebrafish (Roeselers et al., 2011)). The distances were determined using Bray−Curtis method with relative abundance data at phylum level.
The high abundance of Cyanobacteria observed in SVCP and GZSD_F likely support their importance as food sources. Cyanobacteria are known to be important food sources for SVCP (Beveridge et al., 1993) and GZSD (De Brabandere et al., 2009). Within this phylum, Bacillariophyta was the most dominant genus observed in all foregut samples (Figure 2) and has been reported at a high abundance in zebra mussels gut samples (Winters et al., 2011) but not in grass carp (Wu et al., 2012). These observations suggest that Bacillariophyta may be an important diet source for SVCP and GZSD. High abundance of Bacillariophyta genus was also observed in the hindgut of GZSD and SVCP. This suggested that the digestion process occurring in the foreguts of SVCP and GZSD might be incomplete, even though shifts in the microbial community from foregut to hindgut were observed. Kolar et al. (2007) also reported that some Microcystis cells are not killed by the digestive processes of SVCP. Lewin et al. (2003) suspected that Microcystis may pick up nutrients in the gut for growth. Similar incomplete digestion was also observed in young SVCP gut (Henebry et al., 1988). The incomplete digestion in SVCP gut could also be related to a short-residence time, which varied from 4 h at 28.5 °C (Henebry et al., 1988) to 10 h at 20–22 °C (Bialokoz and Krzywosz, 1981). Sharing common Cyanobacteria members further suggest that GZSD and SVCP could compete for the same type of food sources.
No clear patterns based on the NMDS analysis were observed among the microbial communities in foreguts samples. This was not unexpected, as the majority of the foregut content was made up of the food ingested, which included zooplankton, phytoplankton and microbial cells present in the ingested water and zooplankton gut. Also, with a short-residence time in the foregut (Henebry et al., 1988), the development of foregut microbial communities, including those microorganisms involved in cellulose degradation, could be incomplete to effectively degrade the ingested food sources. This possibility was further supported by the observation that microbial populations (for example, Vibrio, Aeromonas, Bacillus, Enterobacter, Anoxybacillus, Leuconostoc, Clostridium, Actinomyces and Citrobacter) previously reported to degrade cellulose (Li et al., 2003; Saha et al., 2006; Wu et al., 2012) were only present at a low abundance in GZSD and SVCP (Table 2). The highest abundance (1.72% of the total sequences) was observed with Clostridium XlVa. The possibility that other novel cellulose-degrading bacteria were present needs to be further confirmed.
The NMDS analysis clearly separated the microflora of GZSD_H and SVCP_H into two different groups, and revealed that the clustering within GZSD_H and SVCP_H samples was strongly affected by sampling locations and sampling time, respectively (Figure 3). A possible explanation could be related to the differences in the swimming behaviors between GZSD and SVCP. GZSD tend to stay at a same location (Miller, 1960), and SVCP usually travel around different locations (Williamson and Garvey, 2005). Using a combination of mobile tracking and stationary receivers, SCVP were recorded to have a daily movement rate of 3.4–64 km d−1 and a maximum travel range of 112–411 km in Illinois river and Swan Lake during Spring−Summer between 2004 and 2005 (DeGrandchamp et al., 2008). Similarly, with the differences in the type of food sources at different locations, GZSD populations in different locations could develop different hindgut microbial communities. This is supported by the observation of Michaletz (1998) that the population characteristics of GZSD were quite variable across different reservoirs. Similar results were also observed among the gut bacterial communities of domesticated zebrafish obtained from aquaculture facilities at different geographical locations (Roeselers et al., 2011). In contrast, SVCP tends to travel a long distance, and this could minimize the variation in the type of food sources ingested at one time point. The effect of sampling time on the SVCP_H composition could suggest that the type of food ingested by SVCP at different seasons is different. However, besides temperature, the seasonal effects attributed to dissolved oxygen and minerals should not be ruled out.
Both GZSD and SVCP are stomachless fish, but can be distinguished by the morphologies of their gastrointestinial tracts. GZSD use gills with filament-like structures to capture food, and have a gizzard to grind ingested food and pyloric caeca to help increase surface area for absorption of nutrients. In contrast, SVCP use gills with a net-like structure and lack a gizzard and phyloric caeca. They mainly use pharyngeal teeth to grind particles before ingestion. These differences could preferentially select food types on the basis of physical size (Sampson et al., 2009) and further shape the microbial communities developed in the hindguts. For this, it was observed that shared OTUs (Figure 4) in different fish gut samples differed significantly in abundances, and unique OTUs in the foreguts and hindguts of SVCP and GZSD could be identified (Figure 4b). Most of the unique OTUs present in GZSD and SVCP gut samples are associated with the phylum Bacteroidetes, one of the most dominant phyla found in the fish gut samples.
Furthermore, a shift in microbial diversity from foregut to hindgut was clearly observed in SVCP and GZSD (Supplementary Figure S5). The shift is likely due to the outcome of the digestion process taking place between the foregut and the hindgut, which is collectively influenced by season, location, dietary input and gut morphology of GZSD and SVCP as discussed above and elsewhere (Sullam et al., 2012). As measurements on key variables such as pH, oxygen concentration, nutritional conditions and digestive enzymes in the foregut and hindgut of individual fish were lacking in this study, it is rather difficult to understand how the digestion process is influenced and regulated, and future study under well-controlled conditions is needed.
In conclusion, GZSD and SVCP were observed to harbor distinct microbial flora in the foregut and hindgut. More than 7000 OTUs were observed in GZSD_H samples, which were higher than other fish gut samples obtained from SVCP (this study), grass carp (Wu et al., 2012) and zebrafish (Roeselers et al., 2011). A change in the microbial diversity between the foregut and hindgut in GZSD and SVCP suggested the influence of food source types and gut morphology on the development of gut microbiota. Hindgut microflora differed between GZSD and SVCP, and microflora of GZSD were affected by location, whereas SVCP_H microflora were affected by sampling time. A few OTUs were observed to be unique or had different abundances in GZSD or SVCP gut flora. Further studies to investigate the microbial composition of gut flora and fecal materials discharged from dominant fish in aquatic environments can provide comprehensive understanding of the differences in diet and physiological behavior among fishes. Moreover, host-specific microbial species for example from the phylum Bacteroidetes can be used to develop potential host-specific molecular monitoring assays for the control of Asian carp and complement existing molecular surveillance techniques for Asian carp.
References
Amberg JJ, Schreier TM, Gaikowski MP . (2012). Molecular responses differ between sensitive silver carp and tolerant bighead carp and bigmouth buffalo exposed to rotenone. Fish Physiol Biochem 38: 1379–1391.
Beveridge M, Baird D, Rahmatullah S, Lawton L, Beattie K, Codd G . (1993). Grazing rates on toxic and non-toxic strains of cyanobacteria by Hypophthalmichthys molitrix and Oreochromis niloticus. J Fish Biol 43: 901–907.
Bialokoz W, Krzywosz T . (1981). Feeding intensity of silver carp (Hypophthalmichthys molitrix Val.) from the Paproteckie Lake in the annual cycle. Ekol Pol 29: 53–61.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK et al (2010). QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7: 335–336.
Chen P, Wiley E, Mcnyset KM . (2007). Ecological niche modeling as a predictive tool: silver and bighead carps in North America. Biol Invasions 9: 43–51.
Chick JH, Pegg MA . (2001). Invasive carp in the Mississippi River basin. Science 292: 2250–2251.
Claesson M, O’Sullivan O, Wang Q, Nikkila J, Marchesi J, Smidt H et al (2009). Comparative analysis of pyrosequencing and a phylogenetic microarray for exploring microbial community structures in the human distal intestine. PLoS One 4: e6669.
Danzeisen JL, Kim HB, Isaacson RE, Tu ZJ, Johnson TJ . (2011). Modulations of the chicken cecal microbiome and metagenome in response to anticoccidial and growth promoter treatment. PLoS One 6: e27949.
De Brabandere L, Catalano M, Frazer T, Allen M . (2009). Stable isotope evidence of ontogenetic changes in the diet of gizzard shad Dorosoma cepedianum. J Fish Biol 74: 105–119.
DeGrandchamp KL, Garvey JE, Colombo RE . (2008). Movement and habitat selection by invasive Asian carps in a large river. Trans Am Fish Soc 137: 45–56.
DeSantis T Jr, Hugenholtz P, Keller K, Brodie E, Larsen N, Piceno Y et al (2006). NAST: a multiple sequence alignment server for comparative analysis of 16S rRNA genes. Nucleic Acids Res 34: W394–W399.
Drake HL, Andreas S, Marcus AH . (2006). Earthworm gut microbial biomes: their importance to soil microorganisms, denitrification, and the terrestrial production of the greenhouse gas N2O. In: Intestinal microorganisms of Termites and Other Invertebrates König H, Varma A (eds). Springer: Berlin, Germany, pp 65-87.
Edgar RC . (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26: 2460–2461.
Hansen SP, Choudhury A, Heisey D, Ahumada J, Hoffnagle T, Cole R . (2006). Experimental infection of the endangered bonytail chub (Gila elegans) with the Asian fish tapeworm (Bothriocephalus acheilognathi): impacts on survival, growth, and condition. Can J Zool 84: 1383–1394.
Henebry M, Gorden R, Buck D . (1988). Bacterial populations in the gut of the silver carp (Hypophthalmichthys molitrix). Prog Fish Cult 50: 86–92.
Herborg LM, Mandrak NE, Cudmore BC, MacIsaac HJ . (2007). Comparative distribution and invasion risk of snakehead (Channidae) and Asian carp (Cyprinidae) species in North America. Can J Fish Aquat Sci 64: 1723–1735.
Hong PY, Hwang C, Ling F, Andersen GL, LeChevallier MW, Liu WT . (2010). Pyrosequencing analysis of bacterial biofilm communities in water meters of a drinking water distribution system. Appl Environ Microbiol 76: 5631–5635.
Hooper LV, Wong MH, Thelin A, Hansson L, Falk PG, Gordon JI . (2001). Molecular analysis of commensal host-microbial relationships in the intestine. Science 291: 881–884.
Huber I, Spanggaard B, Appel K, Rossen L, Nielsen T, Gram L . (2004). Phylogenetic analysis and in situ identification of the intestinal microbial community of rainbow trout (Oncorhynchus mykiss, Walbaum). J Appl Microbiol 96: 117–132.
Hugenholtz P, Goebel BM, Pace NR . (1998). Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J Bacteriol 180: 4765–4774.
Irons KS, Sass G, McClelland M, Stafford J . (2007). Reduced condition factor of two native fish species coincident with invasion of non-native Asian carps in the Illinois River, USA Is this evidence for competition and reduced fitness? J Fish Biol 71: 258–273.
Jerde CL, Mahon AR, Chadderton WL, Lodge DM . (2011). ‘Sight-unseen’ detection of rare aquatic species using environmental DNA. Conserv Lett 4: 150–157.
Kim DH, Brunt J, Austin B . (2007). Microbial diversity of intestinal contents and mucus in rainbow trout (Oncorhynchus mykiss). J Appl Microbiol 102: 1654–1664.
Kolar CS, Chapman DC, Courtenay WR, Housel CM, Jennings DP, Williams JD . (2007) Bigheaded Carps: a Biological Synopsis and Environmental Risk Assessment vol. 33. Amer Fisheries Society: Bethesda, MD, USA.
Krzywinski M, Schein J, Birol İ, Connors J, Gascoyne R, Horsman D et al (2009). Circos: an information aesthetic for comparative genomics. Genome Res 19: 1639–1645.
Lan Y, Verstegen M, Tamminga S, Williams B . (2005). The role of the commensal gut microbial community in broiler chickens. World Poult Sci J 61: 95–104.
Letunic I, Bork P . (2007). Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 23: 127–128.
Lewin W-C, Kamjunke N, Mehner T . (2003). Phosphorus uptake by Microcystis during passage through fish guts. Limnol Oceanogr 48: 2392–2396.
Li X, Dong X, Zhao C, Chen Z, Chen F . (2003). Isolation and some properties of cellulose-degrading Vibrio sp. LX-3 with agar-liquefying ability from soil. World J Microbiol Biotechnol 19: 375–379.
Looft T, Johnson TA, Allen HK, Bayles DO, Alt DP, Stedtfeld RD et al (2012). In-feed antibiotic effects on the swine intestinal microbiome. Proc Natl Acad Sci USA 109: 1691–1696.
McKenna P, Hoffmann C, Minkah N, Aye PP, Lackner A, Liu Z et al (2008). The macaque gut microbiome in health, lentiviral infection, and chronic enterocolitis. PLoS Path 4: e20.
Metzker ML . (2009). Sequencing technologies—the next generation. Nat Rev Genet 11: 31–46.
Michaletz PH . (1998). Population characteristics of gizzard shad in Missouri reservoirs and their relation to reservoir productivity, mean depth, and sport fish growth. N Am J Fish Manage 18: 114–123.
Middelbos IS, Boler BMV, Qu A, White BA, Swanson KS, Fahey GC . (2010). Phylogenetic characterization of fecal microbial communities of dogs fed diets with or without supplemental dietary fiber using 454 pyrosequencing. PLoS One 5: e9768.
Miller RR . (1960). Systematics and biology of the gizzard shad (Dorosoma cepedianum) and related fishes. Fish Bull 60: 371–392.
Ohkuma M, Brune A . (2011). Diversity, structure, and evolution of the termite gut microbial community. Biol Termites Mod Synth, 413–438.
Parks DH, Beiko RG . (2010). Identifying biologically relevant differences between metagenomic communities. Bioinformatics 26: 715–721.
Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C et al (2010). A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464: 59–65.
Ringų E, Olsen RE, Mayhew TM, Myklebust R . (2003). Electron microscopy of the intestinal microflora of fish. Aquaculture 227: 395–415.
Roesch L, Fulthorpe R, Riva A, Casella G, Hadwin A, Kent A et al (2007). Pyrosequencing enumerates and contrasts soil microbial diversity. ISME J 1: 283–290.
Roeselers G, Mittge EK, Stephens WZ, Parichy DM, Cavanaugh CM, Guillemin K et al (2011). Evidence for a core gut microbiota in the zebrafish. ISME J 5: 1595–1608.
Saha S, Roy RN, Sen SK, Ray AK . (2006). Characterization of cellulase-producing bacteria from the digestive tract of tilapia, Oreochromis mossambica (Peters) and grass carp, Ctenopharyngodon idella (Valenciennes). Aquacult Res 37: 380–388.
Sampson SJ, Chick JH, Pegg MA . (2009). Diet overlap among two Asian carp and three native fishes in backwater lakes on the Illinois and Mississippi rivers. Biol Invasions 11: 483–496.
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB et al (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75: 7537–7541.
Shanks OC, Kelty CA, Archibeque S, Jenkins M, Newton RJ, McLellan SL et al (2011). Community structures of fecal bacteria in cattle from different animal feeding operations. Appl Environ Microbiol 77: 2992–3001.
Sullam KE, Essinger SD, Lozupone CA, O’Connor MP, Rosen GL, Knight R et al (2012). Environmental and ecological factors that shape the gut bacterial communities of fish: a meta-analysis. Mol Ecol 21: 3363–3378.
Tamaki H, Wright CL, Li X, Lin Q, Hwang C, Wang S et al (2011). Analysis of 16S rRNA amplicon sequencing options on the Roche/454 next-generation titanium sequencing platform. PLoS One 6: e25263.
Torok VA, Ophel-Keller K, Loo M, Hughes RJ . (2008). Application of methods for identifying broiler chicken gut bacterial species linked with increased energy metabolism. Appl Environ Microbiol 74: 783–791.
Wang Q, Garrity G, Tiedje J, Cole J . (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73: 5261–5267.
Wang T, Cai G, Qiu Y, Fei N, Zhang M, Pang X et al (2011). Structural segregation of gut microbiota between colorectal cancer patients and healthy volunteers. ISME J 6: 320–329.
Welch BL . (1947). The generalization of ‘student’s’ problem when several different population variances are involved. Biometrika 34: 28–35.
Williamson CJ, Garvey JE . (2005). Growth, fecundity, and diets of newly established silver carp in the middle Mississippi River. Trans Am Fish Soc 134: 1423–1430.
Winters AD, Marsh TL, Faisal M . (2011). Heterogeneity of bacterial communities within the zebra mussel (Dreissena polymorpha) in the Laurentian Great Lakes Basin. J Great Lakes Res 37: 318–324.
Wu S, Wang G, Angert ER, Wang W, Li W, Zou H . (2012). Composition, diversity, and origin of the bacterial community in grass carp intestine. PLoS One 7: e30440.
Xu HB, Ge RF, Xiong ML . (1988). Pathogenetic investigation of the enteritis of the grass carp (Ctenopharyngodon idellus). Acta Hydrobiol Sin 12: 308–315.
Ye L, Shao MF, Zhang T, Tong AHY, Lok S . (2011). Analysis of the bacterial community in a laboratory-scale nitrification reactor and a wastewater treatment plant by 454-pyrosequencing. Water Res 45: 4390–4398.
Zhang C, Zhang M, Wang S, Han R, Cao Y, Hua W et al (2009). Interactions between gut microbiota, host genetics and diet relevant to development of metabolic syndromes in mice. ISME J 4: 232–241.
Acknowledgements
We thank Ya Zhang, Na Kyung Kim, Fangqiong Ling, Chiachi Hwang and Hideyuki Tamaki for technical assistance. This work was funded by a collaborative grant through United States Geological Survey. We also thank the editor and two anonymous reviewers for their constructive comments on the original submission. Last, we would like to thank Duane Raver, Jr for providing original illustrations of SVCP and GZSD used in Figure 1.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article
Cite this article
Ye, L., Amberg, J., Chapman, D. et al. Fish gut microbiota analysis differentiates physiology and behavior of invasive Asian carp and indigenous American fish. ISME J 8, 541–551 (2014). https://doi.org/10.1038/ismej.2013.181
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2013.181
Keywords
This article is cited by
-
Influence of gut microbiome on health and development of penaeid shrimps
Aquatic Sciences (2024)
-
Effect of two insect meals on the gut commensal microbiome of healthy sea trout (Salmo trutta vr. trutta)
BMC Veterinary Research (2023)
-
In silico prediction of potential indigenous microbial biomarkers in Penaeus vannamei identified through meta-analysis and genome-scale metabolic modelling
Environmental Microbiome (2023)
-
The effects of host quantitative genetic architecture on the gut microbiota composition of Chinook salmon (Oncorhynchus tshawytscha)
Heredity (2023)
-
Starvation alters gut microbiome and mitigates off-flavors in largemouth bass (Micropterus salmoides)
Folia Microbiologica (2023)
