
Abstract
Dimethylsulfoniopropionate (DMSP) is an important source of reduced sulfur and carbon for marine microbial communities, as well as the precursor of the climate-active gas dimethylsulfide (DMS). In this study, we used metatranscriptomic sequencing to analyze gene expression profiles of a bacterial assemblage from surface waters at the Bermuda Atlantic Time-series Study (BATS) station with and without a short-term enrichment of DMSP (25 nM for 30 min). An average of 303 143 reads were obtained per treatment using 454 pyrosequencing technology, of which 51% were potential protein-encoding sequences. Transcripts from Gammaproteobacteria and Bacteroidetes increased in relative abundance on DMSP addition, yet there was little change in the contribution of two bacterioplankton groups whose cultured members harbor known DMSP degradation genes, Roseobacter and SAR11. The DMSP addition led to an enrichment of transcripts supporting heterotrophic activity, and a depletion of those encoding light-related energy generation. Genes for the degradation of C3 compounds were significantly overrepresented after DMSP addition, likely reflecting the metabolism of the C3 component of DMSP. Mapping these transcripts to known biochemical pathways indicated that both acetyl-CoA and succinyl-CoA may be common entry points of this moiety into the tricarboxylic acid cycle. In a short time frame (30 min) in the extremely oligotrophic Sargasso Sea, different gene expression patterns suggest the use of DMSP by a diversity of marine bacterioplankton as both carbon and sulfur sources.
Similar content being viewed by others
Introduction
Dimethylsulfoniopropionate (DMSP) is a reduced organic sulfur compound produced by many species of phytoplankton in the euphotic zone of the oceans (Keller et al., 1989). At the global scale, this compound can supply most of the sulfur and part of the carbon requirements of marine microbial communities (Kiene and Linn, 2000a; Simó et al., 2009). Furthermore, the degradation product dimethylsulfide (DMS) represents a major biogenic flux of sulfur from oceans to land through the atmosphere (Lovelock et al., 1972). Atmospheric DMS-oxidation products can influence climate through backscattering of incoming solar radiation, promoting a cooling effect on the planet (Charlson et al., 1987; Simó, 2001).
Bacteria are one of the main mediators of the fate of DMSP in seawater. When DMSP is used as a sulfur and carbon source, it is demethylated to form methylmercaptopropionate (MMPA), which can be demethiolated to methanethiol (MeSH) and used in amino-acid synthesis. Alternatively, DMSP can be cleaved to produce DMS and a C3 compound (Kiene and Linn, 2000b; Todd et al., 2007). Cultured bacteria that are able to carry out both pathways have been described (González et al., 1999; Yoch, 2002), and assimilation of the sulfur moiety appears to be a widespread capability in natural bacterial communities (Yoch, 2002; Malmstrom et al., 2004a; Vila-Costa et al., 2007). However, for most bacterial transformations involved in DMSP degradation, the identity of the main bacterial groups, and the factors controlling the biological switch between DMSP pathways, remain poorly understood.
Recently, genes encoding the first step of DMSP degradation have been identified in marine bacterial isolates. The gene dmdA, identified in Ruegeria pomeroyi DSS-3, encodes for the demethylation of DMSP (Howard et al., 2006). Homologs to this gene are present in up to 58% of cells sampled in the Global Ocean Sampling (GOS) metagenome. On the basis of the similarity to homologs in cultured bacteria, most GOS sequences appear to be harbored by Alphaproteobacteria, with a small contribution from oligotrophic Gammaproteobacteria (Howard et al., 2008). Three genes encoding the first step in the competing DMS production pathway have also been identified. dddL encodes a DMSP lyase that cleaves DMSP to DMS (Curson et al., 2008). In contrast, dddD encodes an acyl-CoA transferase that converts DMSP to 3-OH-propionate and DMS (Todd et al., 2007, 2010). Finally, dddP encodes a peptidase that converts DMSP to DMS through an unknown mechanism (Todd et al., 2009). Relative to the DMSP demethylase gene, the DMSP cleavage genes are underrepresented in GOS, averaging only 1.4% of cells (0.2% for dddD, 0.6% for dddL, 3.5% for dddP; Howard et al., 2008; Todd et al., 2009). These recent molecular studies are paving the way to an improved understanding of the abundance, expression and regulation of DMSP-related genes in seawater.
Metatranscriptomics, the sequencing of messenger RNA (mRNA, representing expressed genes) from the environment, provides an unbiased view of the functionality of a microbial community as it is not constrained to targeting a specific phylogenetic group or metabolic pathway (Poretsky et al., 2005). To this point, metatranscriptomic analyses have shown community expression patterns over diel cycles or under enhanced CO2 concentrations, as well as identified taxon-specific metabolic processes (Frias-Lopez et al., 2008; Gilbert et al., 2008; Hewson et al., 2009; Poretsky et al., 2009). The technique is particularly amenable to experimental manipulations designed to test hypotheses about complex microbial communities (Gilbert et al., 2008; Moran, 2008, 2009; Poretsky et al., 2010).
The Bermuda Atlantic Time-series Study (BATS) site is located in the subtropical gyre of the Atlantic Ocean, in the northwest corner of the Sargasso Sea. It is characterized by nutrient-depleted waters in the euphotic zone broken by upwelling eddies that increase chlorophyll a (Chla) in the surface during spring (McGillicuddy, 1998). Three characteristics make this location ideal for genomic-based DMSP studies: (1) DMSP dynamics and pools have been described in previous studies and point to an active role for bacteria in the cycle (Ledyard and Dacey, 1996; Dacey et al., 1998; Harada et al., 2004); (2) a high percentage of bacteria assimilate DMSP in this location (Malmstrom et al., 2004b, 2005); and (3) an extensive metagenomic database already exists for this site (Venter et al., 2004; although from a different date than our study).
In this study, we enriched water from the Sargasso Sea with low amounts of DMSP (25 nM; fivefold above ambient) and compared gene expression with that in a no-addition control. The goals of this study were to: (1) characterize bacterioplankton transport and metabolism related to DMSP processing; (2) identify the main players of DMSP consumption; and (3) identify, if possible, additional genes involved in DMSP degradation.
Materials and methods
Sample collection and enrichment
Sampling was carried out at the Bermuda Atlantic Time-series Study (BATS) station (31° 40. 1′N, 64° 7.9′W) at a depth of 10 m using 10-l Niskin bottles on 15 April 2008. A subsample of 2 l of unfiltered water was collected to measure in situ concentrations of Chla and total DMSP. To exclude eukaryotes and large particles, the remaining seawater was prefiltered by gravity through 3 μm pore-size polycarbonate filters (142 mm, Millipore, Billerica, MA, USA) using an inlet filtration system attached directly to the Niskin bottle. Water was dispensed into 20 l polycarbonate carboys and maintained in a temperature-controlled room in the dark at in situ temperature for 3 h before beginning the experiment to allow time for adaptation to any DMSP released from the cells during filtration.
DMSP was synthesized from DMS and acrylate, and determined to be >98% pure by 1H nuclear magnetic resonance analysis. To begin the experiment, DMSP was added to experimental carboys to a final concentration of 25 nM and control carboys remained untreated. After 30 min, the bacterial cells were collected by filtering the water through 0.2 μm pore-size polycarbonate filters (142 mm diameter) using a peristaltic pump. The duration of the filtration step was no longer than 20 min to minimize RNA degradation. Filters were placed in 15 ml RNAse-free tubes containing 2 ml of Buffer RLT (RNeasy kit, Qiagen, Valencia, CA, USA) plus 10 μl of β-mercaptoethanol per ml. Tubes were flash-frozen in liquid nitrogen and stored at −80 °C until further processing.
An additional control carboy was used for DNA extraction for 16S rRNA community analysis, following the same procedure except that no RLT buffer was added. Filters were placed in sterile plastic bags (Whirlpak, Nasco, Fort Atkinson, WI, USA), flash-frozen in liquid nitrogen and extracted later in the laboratory. Chla, dissolved DMSP (DMSPd) concentrations and bacterial production were measured using standard methods as described in the Supplementary Material.
RNA preparation
Messenger RNA (mRNA) extraction, enrichment, amplification, and conversion to complementary DNA was performed as described by Poretsky et al. (2009) with few modifications (see Supplementary Material). Only one replicate from the experimental and control treatments yielded RNA of sufficient quality for processing.
Pyrosequencing
Complementary DNA libraries were sequenced with Roche GS FLX sequencing (Branford, CT, USA), yielding 606 286 reads (209-bp average length) (Supplementary Table S1). The sequences have been deposited according to MIMS standards in the Community Cyberinfrastructure for Advanced Marine Microbial Ecology Research and Analysis (CAMERA) database (http://www.camera.calit2.net/index.php) with the Genome Project ID CAM_PROJ_SargassoSea.
rRNA identification
Exact duplicate reads (10.7% of total reads) were removed from the libraries. Ribosomal RNA reads were identified by performing a BLASTn search against the rRNA Silva database (http://www.arb-silva.de/) with a cut-off criterion of E-value ⩽10−5. The rRNA reads accounted for 49% of the total reads (81.7% with best hits to Bacteria, 10.2% to Eukarya and 0.2% to Archaea) and were not considered further.
Complementary DNA sequence annotation
Potential protein-encoding reads were identified by querying remaining reads with BLASTx against the RefSeq database (NCBI) with cut-off criteria of E-value ⩽10−2, amino-acid alignment length ⩾23, and similarity >40% (established previously with in silico tests of known genes; see Mou et al., 2008). Identified proteins were classified on the basis of the COG (Tatusov et al., 2003) and KEGG (Kanehisa and Goto, 2000) databases with cut-off criteria of E-value ⩽0.1, amino-acid alignment length ⩾23, and similarity >40% for both databases (see Mou et al., 2008). Transcripts with no matches in RefSeq were BLASTx-queried against the NCBI nr database and against the CAMERA database of unassembled ORFs predicted from the GOS reads (Seshadri et al., 2007). Taxonomic affiliation of identified transcripts was assigned to the order level using MEGAN (Huson et al., 2007) with default settings for version 2.0.
Identification of homologs to reference genes
Known query genes related to DMSP and sulfur (S) metabolism were selected from the literature, using orthologs in three genomes representing major marine bacterioplankton groups (Pelagibacter ubique SAR11, Ruegeria pomeroyi DSS-3 and Gammaproteobacterium HTCC 2080) whenever possible (Supplementary Table S2). These DMSP- and S-related protein sequences were used in BLASTx analysis of the Sargasso Sea transcripts to identify reads potentially encoding DMSP-related functions (cut-off criterion of E-value <10−2). Hits in the transcript libraries were analyzed by BLASTx against the RefSeq database (NCBI) and designated an ortholog if the top hit was to a gene of the correct function with a cut-off criterion of bit-score >50. The validity of the annotation criteria was checked by manual annotation of dmdA hits.
Statistical analysis
Significantly overrepresented functional categories (COGs, KEGGs or DMSP-related genes) in the transcript libraries were identified with the statistical software Xipe at varying confidence levels (Rodriguez-Brito et al., 2006). Because sequencing was restricted to one experimental and one control carboy, there were no within-treatment analyses of variability. The program was run with 20 000 resamplings for a sample size of 39 000 for COGs, 22 000 for KEGGs and 1200–4500 (depending on the number of genes in the library) for DMSP gene categories.
DNA preparation and sequencing
DNA was extracted using PowerMax Soil Mega Prep DNA Isolation kit (MoBio, Carlsbad, CA, USA). Partial bacterial 16S ribosomal RNA genes that included the hypervariable region V6 were amplified by PCR using a mix of five forward primers (967F) and four reverse primers (1046R; Sogin et al., 2006). The reaction was carried out with Ready-To-Go PCR beads (Amersham Pharmacia, Piscataway, NJ, USA) with a final concentration of forward and reverse primer mixtures of 0.1 μM. PCR conditions were as follows: 5 min at 94 °C, followed by 30 cycles of denaturation at 94 °C for 1 min, annealing at 55 °C for 1 min, primer extension at 72 °C for 1 min, and a final extension at 72 °C for 10 min. PCR products were cleaned using the QIAquick PCR Purification kit (Qiagen) and the Agencourt AMPure XP kit (Agencourt Bioscience, Beckman Coulter, Brea, CA, USA). Amplicons were sequenced in a partial Roche GS FLX run yielding 21 049 reads (63 bp average length after primer removal). Amplicons were clustered at 99% similarity using the CD-Hit package (Li and Godzik, 2006). Taxonomic affiliation of the 99% clusters was determined by pairwise Smith-Waterman identity calculations of the reference sequences to members of an in-house database containing marine 16S rRNA bacterial and archaeal gene sequences retrieved from the Ribosomal Database Project (RDP) and the GOS database (Biers et al., 2009; the reference database is available at http://www.simo.marsci.uga.edu/public_db/bioinformatics.htm). A minimum of 90% sequence identity with an overlap of 80% of the input sequence length was required for taxonomic assignment.
Results
Environmental parameters
Seawater temperature was 21.2 °C, salinity was 36.7 and Chla concentration was 0.028 μg l−1 in the initial whole water. The total DMSP concentration was 18.3±1.2 nM, with over 80% of this also detected in the <3-μm size fraction (15.4±4.7 nM), indicating that most DMSP producers were pico- and nanoeukaryotes. Dissolved DMSP concentration was 4.9±1.8 nM at the initial time point, and remained constant in the control treatment until RNA was collected (5.4±2.3 nM). The 25 nM addition of DMSP to the experimental treatment resulted in elevated DMSPd concentrations, measured as 27.8±1.6 nM just before RNA collection. Bacterial production was 1.2±0.2 nM leucine per day in the initial <3 μm size fraction, 1.0±0.2 nM per day in the control treatment after a 3 h incubation, and 1.1±0.2 nM per day in the experimental treatment after a 3 h incubation. Bacterial production incubations were longer than gene expression incubations (3 h vs 30 min) to allow for detectable radioisotope uptake in this oligotrophic environment.
Overall patterns in gene expression
An average of 303 143 sequence reads were obtained per library, with a mean length of 209 bp (Supplementary Table S1). Our methodology yielded 51% potential protein-encoding sequences, of which approximately half (∼24% of the total sequences) had similarity to protein sequences in the NCBI RefSeq database (see materials and methods section for BLAST criteria). An average of 15% of the total reads could be assigned to a COG category and 22% to a KEGG pathway. Sequences unidentified in RefSeq but with similarity to sequences in the GOS metagenome accounted for 9% of total reads (Supplementary Table S1). An average of 17% of sequences remained unidentified, possibly representing unknown genes, poorly conserved regions of known genes (Poretsky et al., 2010) or small RNAs with regulatory functions (Shi et al., 2009).
In general, functional assignments of transcripts in control versus experimental treatments indicated that bacterioplankton activity was stimulated within 30 min of DMSP addition. COG and KEGGs categories related to transcription and translation were significantly enriched in the transcript pool after DMSP addition according to a resampling-based statistical comparison (98% confidence level; Rodriguez-Brito et al., 2006) (Figure 1, Supplementary Tables S3, S4). Bacterial production measurements based on 3H-leucine uptake did not yet show statistically higher rates of protein synthesis following DMSP amendment (see above), and there was no indication of increases in mRNAs directing DNA replication or cell division (Figure 1, Supplementary Tables S3, S4). These results are in accordance with models for shifts up in bacterial activity, in which RNA is the first macromolecule pool to respond in advance of increased protein synthesis and cell division (Ingraham et al., 1983; Chin-Leo and Kirchman, 1990).

Relative abundance of Sargasso Sea bacterioplankton transcripts in the most abundant COG functional categories.
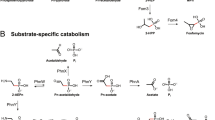
DMSP addition also resulted in an enrichment of transcripts related to both propanoate metabolism and the tricarboxylic acid cycle. These may represent a common set of genes used for the degradation of the 3-carbon (C3) moiety of DMSP (Figure 2, Supplementary Figure S1, and Supplementary Table S5). A gearing-up of metabolism in response to DMSP was also suggested by enrichment of the COG category for energy production (functional category C) (Figure 1), as well as genes related to adenosine triphosphate synthesis, amino-acid metabolism and lipid metabolism (Figure 3, Supplementary Table S4).

Possible degradation pathways of the C3 moiety of DMSP, on the basis of the KEGG pathway 00640. Gray filled rectangles indicate enzymes for which transcripts were enriched after DMSP addition. White rectangles with black frames indicate enzymes for which transcripts were depleted after DMSP addition. Statistical significance (P<0.05) is indicated with an asterisk. A possible major route of degradation of the C3 moiety from propanoate to acetyl-CoA is highlighted in black, based on high numbers of transcripts for most steps.

Relative abundance of transcripts for DMSP-related functional gene categories (C3: acrylate degradation, dddD and dddP: DMS production, dmdA: DMSP demethylation, DMSox: DMS oxidation, MeSH: methanthiol incorporation, TRANS: DMSP uptake system, DMSORED: DMSO reduction, soxB: inorganic sulfur oxidation, Sulfate: assimilatory sulfate reduction, MSA: methanesulfonic acid monooxygenase, MICRO: upregulated genes in a roseobacter-based microarray after DMSP addition, GBT: glycine betaine degradation), and other selected gene categories (inset; PROT: proteorhodopsin, adenosine triphosphate (ATP): ATP synthase). For more information see Supplementary Table S2. The C or D at the end of the category names refers to the Control or DMSP libraries, respectively. Categories statistically enriched or depleted in the DMSP library are labeled with an asterisk (P<0.05). Taxonomy is shown at the greatest level of detail assigned by MEGAN, and there is no overlap between categories. Some abundant Alphaproteobacteria groups are shown to family level (Rhodobacterales, which includes Roseobacters, and Rickettsiales, which includes SAR11).
Transcript categories that were depleted after DMSP addition included bacterial and archaeal genes mediating photo-dependent metabolisms such as photosynthesis and proteorhodopsin-based energy acquisition (Supplementary Tables S3, S4, Figure 3). Also depleted were transcripts for chaperone-related activities (Figure 1, Supplementary Tables S3, S4), with 10 of the 18 individual significantly underrepresented COGs corresponding to proteases and chaperones (functional category O; Supplementary Table S4). DMSP addition reduced the relative abundance of ABC transporter sequences in the community transcriptome (Supplementary Tables S3, S4) as well as expressed genes related to cell motility (Figure 1, Supplementary Table S3, COG functional category N in Supplementary Table S4). These differences are consistent with the hypothesis that bacterioplankton were limited by the availability of labile carbon, with solar radiation providing more of the cells’ energy and protein recycling having a more important role in cellular activity before the addition of DMSP. Because transcript dynamics are evaluated as a percent of the total community transcriptome, data indicate a difference in the relative, not absolute, abundance of any given transcript category in response to DMSPd addition.
Expression of known DMSP-related genes
DMSP-related genes were selected from the literature and queried against both transcriptomes using BLAST. As few bacterial genes known to be directly involved in DMSP degradation have been identified (DMSP demethylation, DMS production and DMS oxidation gene categories; Supplementary Table S2), other genes identified in the genome of model organism Ruegeria pomeroyi DSS-3 (Moran et al., 2004), upregulated in a microarray study of DMSP degradation by R. pomeroyi (Gifford et al., in preparation), potentially involved in DMSP metabolite degradation (MeSH incorporation; Kiene et al., 1999), and from related metabolisms (inorganic S-oxidation, assimilatory sulfate reduction, methanesulfonic acid monooxygenase, glycine betaine degradation) were also included (Supplementary Table S2). The DMSP-related query genes were grouped into categories based on their possible role in DMSP processing (Figure 3).
The DMSP-related transcripts identified by the BLAST analysis were significantly enriched in the experimental transcriptome compared with the control (the combination of all gene categories shown in Figure 3). The subset of DMSP-related transcripts linked to the degradation of the C3 moiety of DMSP through acrylate or propanoate (Figure 3) was individually significant (Xipe analysis; P=0.05).
Taxonomic analysis
Transcripts that binned to Gamma-proteobacteria and Bacteroidetes genes were enriched in the DMSP treatment compared with the non-amended control (Supplementary Figure S2), yet several taxa with known links to DMSP metabolism, such as Roseobacter and SAR11 (Moran et al., 2004; Howard et al., 2008; Tripp et al., 2008), were not. Transcripts from photosynthetic bacteria Synechoccocus and Prochloroccocus were underrepresented in the DMSP transcriptome, although their capacity to assimilate DMSP-S in seawater has been shown in previous studies (Malmstrom et al., 2005; Vila-Costa et al., 2006) (Supplementary Figure S2). This decrease is consistent with the general depletion of transcript categories mediating photosynthesis after addition of DMSP (Supplementary Table S3).
For KEGG pathways and DMSP-related transcript categories significantly enriched after DMSP addition, the same taxonomic pattern was observed. Gammaproteobacteria- and Bacteroidetes-like transcripts were commonly mapped to propanoate and tricarboxylic acid cycle KEGG pathways (Supplementary Figure S3). The transcripts with homology to DMSP-related genes (Figure 3) were assigned to a wide range of taxonomic groups. Rhodobacterales-like transcripts and those from other Alphaproteobacteria groups accounted for the greatest fraction of the DMSP-related transcript pools, consistent with the phylogeny of the previously identified DMSP genes in isolates (Supplementary Table S2).
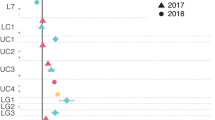
Community composition assessed by taxonomic assignments of PCR amplicons of the V6 region of 16S rRNA genes from community DNA showed a dominance of members of the SAR11 taxon (∼35% of sequences). Although abundant in 16S rRNA amplicon libraries (assuming no major biases in PCR amplifications), this group appears to have low per-cell levels of gene expression and did not show a rapid transcriptional response (that is, within the 30 min incubation) to DMSP addition (Figure 4). This finding is consistent with previous marine metatranscrioptomic studies (Hewson et al., 2009; Poretsky et al., 2009, 2010) and suggests that SAR11 members may not exhibit substantial transcriptional responses to substrate supply, at least on short time frames. Gammaproteobacteria and Alphaproteobacteria other than SAR11 and Roseobacters were the next most abundant groups in the 16S rRNA library (∼20% each), dominated by SAR86 (7%) and SAR116 (8%). Roseobacter-like sequences accounted for less than 5% of 16S rRNA amplicons but more than 10% of the transcripts for both treatments (Figure 4), suggesting higher per-cell levels of gene expression relative to the average cell. Similarly, Bacteriodetes-like sequences accounted for 6% of the 16S rRNA amplicons and up to 11% of the transcripts, with higher per-cell expression after DMSP addition (Figure 4).

Contribution of each phylogenetic group to the 16S rRNA amplicon pool versus the community transcript pool. Dots at the origin represent the Planctomycetes (1), Delta-/Epsilonproteobacteria (2), and Actinobacteria (3). Open circles indicate groups contributing differentially to the transcript pool following DMSP addition. The dotted lines indicate equal contributions to the 16S rRNA amplicon and transcript libraries. Abundance of 16S rRNA gene amplicons may have been influenced by PCR biases, but these are expected to be consistent between control and DMSP treatments.
Discussion
Metatranscriptomics has no requirement for a preselected diagnostic gene set represented as probes or primers, and makes no prior assumptions of the metabolisms present in a community. It therefore provides a more expansive view of the metabolic and taxonomic basis for microbial activities compared with prevailing methods such as PCR assays, microarray approaches and single-cell techniques that target specific genes, taxa or processes. In this study, the rapid enrichment of transcripts mediating degradation of propanoate-like compounds and tricarboxylic acid cycle activity from Sargasso Sea bacterioplankton groups not known to harbor orthologs to DMSP demethylation (Howard et al., 2008) or cleavage genes (Todd et al., 2007, 2010; Curson et al., 2008), provides evidence for the involvement of a greater number of marine taxa in DMSP degradation. This agrees with previous findings for DMSP-S assimilation using single-cell techniques (Vila et al., 2004; Malmstrom et al., 2004a), and suggests that Bacteroidetes, Betaproteobacteria and other bacterioplankton groups either have orthologs of known DMSP-degrading genes that are not being correctly assigned, or have non-homologous genes with similar functions in DMSP degradation that have yet to be found. It is worthy to note that some of the more informative changes in transcript profiles in this experimental manipulation of a marine bacterioplankton community were from genes that are shared by several metabolic pathways and common to many bacteria, not from those indicative of a specific biogeochemical process. Accordingly, these genes are not likely to have been included in preselected diagnostic gene sets for use with more targeted methodologies.
An important advantage of the experimental design was that the 30 min incubation time limited the possibility for uptake of DMSP metabolites by non-DMSP-degrading bacteria. For other methodologies that have been developed to link microbial activities to specific taxa, such as MAR-FISH (Vila et al., 2004; Malmstrom et al., 2004a) and stable isotope probing (Neufeld et al., 2007), longer incubation times are required. However, the short incubation time may have biased transcript retrieval against oligotrophic taxa that respond slowly to changing environmental conditions, such as SAR11 cells that have few regulatory systems for environmental cues (Giovannoni et al., 2005). Conversely, some taxa that responded rapidly to DMSP increases, such as Bacteroidetes and Gammaproteobacteria, may not dominate DMSP degradation under typical conditions (DMSP concentrations <5 nM; Kiene and Slezak, 2006), although they could be important when DMSP supply is higher, such as during phytoplankton blooms or in phycospheres.
DMSP is typically considered a preferred source of sulfur (S) for marine microorganisms (Simó, 2001), as it can be demethylated and then demethiolated to MeSH and used to synthesize methionine and cysteine (Kiene et al., 2000). Although sulfate is much more abundant than DMSP in seawater (mM concentrations instead of nM), there are energetic advantages to taking up S that is already reduced. Previous studies have shown that DMSP can satisfy almost all the microbial community sulfur demands (Kiene and Linn, 2000a; Simó et al., 2002; Zubkov et al., 2002), and is particularly important in oligotrophic environments during periods of high solar irradiation when it contributes more to S fluxes (Simó et al., 2009). Accordingly, DMSP-assimilating cells can account for up to 60% of the community in the Sargasso Sea (Vila et al., 2004; Malmstrom et al., 2004a, 2004b). Kiene et al. (1999) hypothesized that the enzyme cystathionine gamma-synthase (metB) could be responsible for methionine synthesis from DMSP by O-acyl homoserine based on studies with cultured roseobacters, with cysteine subsequently synthesized from methionine. Experimental evidence with isolates of SAR11 that lack genes for the assimilatory sulfate reduction pathway agrees that synthesis of methionine followed by conversion to cysteine might be a common pathway for DMSP-S assimilation, as cysteine itself was unable to support SAR11 growth (Tripp et al., 2008). In contrast, we found that transcripts related to the cysteine synthesis pathway (cysteine synthase (COG0031) and O-acetylhomoserine sulfhydrylase (COG2873, Supplementary Table S4)) were significantly enriched after DMSP addition. Simultaneous increases in gamma-glutamylcysteine synthetase transcripts (COG2918, Supplementary Table S4) involved in glutathione synthesis from cysteine suggests that redox changes in the cell during DMSP degradation might be an alternate explanation for the rapid expression signal from the cysteine synthesis pathway. In any case, the major pathway used by marine bacteria to synthesize amino acids from DMSP-S did not clearly emerge from this analysis.
Both the DMSP demethylation and cleavage pathways yield a C3 compound, possibly acrylate (Kiene and Linn, 2000b), although recent studies suggest that acryloyl-CoA or 3-OH-propionate can also be intermediates (Curson et al., 2008; Todd et al., 2010). Studies of marine and freshwater isolates have shown that some bacteria can grow on acrylate by conversion to 3-OH-propionate (Kiene, 1990; Ansede et al., 1999, 2001; Yoch, 2002). The pattern of transcript enrichment after DMSP addition suggests that the most common degradation pathway of the C3 moiety of DMSP is to acetyl-CoA by malonyl-CoA (Figure 2, see abundances of propanoate-related transcripts in Supplementary Table S5), although this is likely to be taxon-dependent and possibly temporally variable. Studies of cultured DMSP-degrading marine strains suggest that roseobacters degrade C3 compounds derived from DMSP cleavage to succinyl-CoA through the methylmalonyl pathway (C Reisch and S Gifford, personal communication), whereas a marine gammaproteobacterium strain cleaves DMSP to acetyl-CoA through 3-OH-propionyl-CoA (Todd et al., 2010). All these pathways are possible and may co-occur in seawater. Indeed, the enrichment pattern of propanoate-related transcripts suggests substantial metabolic diversity of bacterial DMSP degraders.
The DMSP-induced depletion of transcripts mediating phototrophy (photosynthesis and proteorhodopsins; Supplementary Table S3, Figure 3) concomitant with evidence for gearing-up of heterotrophic metabolism (Supplementary Table S3) suggests that the in situ bacterioplankton community was limited by the availability of labile organics. Although the ecological role of proteorhodopsins in seawater is still unclear (Furhman et al., 2008), they work as light driven pumps that establish a proton gradient across the cell membrane (Beja et al., 2000). Among the broad array of suggested ecological roles of proteorhodopsin (Furhman et al., 2008), our findings are consistent with it functioning as a source of energy under oligotrophic conditions, allowing more efficient bacterial consumption of the available organic carbon (Gómez-Consarnau et al., 2007; Furhman et al., 2008). However, we point out that because our analyses are based on relative values only (that is, transcript abundance as a percent of the total), constant per-cell expression of PR genes in parallel with increased expression of other genes would deplete PR representation as a percent of the transcript pool.
DMSP-related transcripts showed a general dominance of sequences binning to Alphaproteobacteria genomes, particularly roseobacters (Figure 3). Although in low abundance in this oligotrophic ecosystem, the group has accounted for a large proportion of DMSP metabolism in previous studies in oligotrophic sites (Malmstrom et al., 2004a; Vila-Costa et al., 2008). SAR11 members were the most abundant taxon in the bacterial assemblage based on PCR-amplified 16S rRNA genes (Figure 4), but were poorly represented in the DMSP-related transcript pool. As the ability of SAR11 members to assimilate DMSP has been shown (Malmstrom et al., 2004b; Tripp et al., 2008), they may have responded more slowly than other groups to the DMSP addition, or the concentration used here (25 nM compared with typical DMSPd concentrations of 3–6 nM; Kiene and Slezak, 2006) might have saturated their transport systems. The fact that DMSP concentrations were still high in the experimental treatment at the time of RNA collection for sequencing (28 nM) suggests that bacterioplankton transcriptional responses should be ongoing at the time of sample collection. Transcripts binning to Gammaproteobacteria and Bacteroidetes genes increased after DMSP addition (Supplementary Figure S2), with the same pattern observed for just that subset of transcripts related to C3 degradation (Supplementary Figure S2). Gamma-proteobacteria often account for a large proportion of the DMSP assimilating bacterial population in seawater (Vila et al., 2004; Malmstrom et al., 2004a; Vila-Costa et al., 2007), but Bacteroidetes are thought to prefer high-molecular-weight organic molecules (Cottrell and Kirchman, 2000). A ‘bottle effect’ that enhanced Gammaproteobacterial activity levels above what is typical for the Sargasso Sea cannot be ruled out, as this group is classically considered opportunistic (Massana et al., 2001). The low concentration of added DMSP and short incubation time likely minimized this effect, but in any case does not alter the finding that Gammaproteobacteria are capable of responding rapidly to 25 nM DMSP. Synechoccocus and Prochlococcocus have been shown previously to compete with heterotrophic bacteria for DMSP (Malmstrom et al., 2005; Vila-Costa et al., 2006), but we saw no increase of their transcriptional activity in the experimental treatment; light was required to stimulate DMSP uptake in these earlier studies.
Diverse bacterioplankton taxa in the oligotrophic Sargasso Sea responded to a DMSP addition in a very short time frame (30 min), suggesting the use of DMSP by a large number of marine bacteria as either C or S sources, or both. The C3 moiety of DMSP is degraded by the community through propanoate-like compounds, and enters the tricarboxylic acid cycle by a number of pathways. The response to DMSP addition in downstream pathways by Bacteroidetes and phylogenetic groups other than Gamma- and Alpha-proteobacteria suggests that there are important DMSP degradation genes yet to be identified. Although this study highlighted the activities of the most rapidly responding sector of the bacterial community, a metatranscriptomic time series over hours to days could capture the activity of more slowly responding cells. This functional metagenomics approach to bacterial DMSP degradation represents a first view of the diversity of the metabolisms operating during DMSP degradation in the open ocean.
References
Ansede JH, Pellechia PJ, Yoch DC . (1999). Metabolism of acrylate to β-hydroxypropionate and its role in dimethylsulfoniopropionate lyase induction by salt marsh sediment bacterium, Alcaligenes faecalis M3A. Appl Environ Microbiol 65: 5075–5081.
Ansede JH, Pellechia PJ, Yoch DC . (2001). Nuclear magnetic resonance analysis of [1-13C]dimethylsulfoniopropionate (DMSP) and [1-13C]acrylate metabolism by a DMSP lyase-producing marine isolate of the α-subclass of proteobacteria. Appl Environ Microbiol 67: 3134–3139.
Beja O, Aravind L, Koonin EV, Suzuki MT, Hadd A, Nguyen LP et al. (2000). Bacterial rhodopsin: evidence for a new type of phototrophy in the sea. Science 289: 1902–1905.
Biers EJ, Sun S, Howard EC . (2009). Prokaryotic genomes and diversity in surface ocean waters: interrogating the global ocean sampling metagenome. Appl Environ Microbiol 75: 2221–2229.
Charlson RJ, Lovelock JE, Andreae MO, Warren SG . (1987). Oceanic phytoplankton, atmospheric sulfur, cloud albedo and climate. Nature 326: 655–661.
Chin-Leo G, Kirchman DL . (1990). Unbalanced growth in natural assemblages of marine bacterioplankton. Mar Ecol Prog Ser 63: 1–8.
Cottrell MT, Kirchman DL . (2000). Natural assemblages of marine protebacteria and members of the Cytophaga-Flavobacter cluster consuming low- and high-molecular-weight dissolved organic matter. Appl Environ Microbiol 66: 1692–1697.
Curson ARJ, Rogers R, Todd JD, Brearley CA, Johnston AWB . (2008). Molecular genetic analysis of a dimethylsulfoniopropionate lyase that liberates the climate-changing gas dimethylsulfide in several marine alpha-proteobacteria and Rhodobacter sphaeroides. Environ Microbiol 10: 757–767.
Dacey JWH, Howse FA, Michaels AF, Wakeham SG . (1998). Temporal variability of dimethylsulfide and dimethylsulfoniopropionate in the Sargasso Sea. Deep-Sea Res 45: 2085–2104.
Frias-Lopez J, Shi Y, Tyson GW, Coleman ML, Schuster SC, Chisholm SW et al. (2008). Microbial community gene expression in ocean surface waters. Proc Nat Acad Sci 105: 3805–3810.
Furhman JA, Schwalbach MS, Stingl U . (2008). Proteorhodopsins: an array of physiological roles? Nat Rev Microbiol 6: 488–494.
Gilbert JA, Field D, Huang Y, Edwards R, Li W, Gilna P et al. (2008). Detection of large numbers of novel sequences in the metatranscriptomes of complex marine microbial communities. PLoS ONE 3: e3042.
Giovannoni SJ, Tripp HJ, Givan S, Podar M, Vergin KL, Baptista D et al. (2005). Genome streamlining in a cosmopolitan oceanic bacterium. Science 309: 1242–1245.
Gómez-Consarnau L, Gonzalez JM, Coll-Llado M, Gourdon P, Pascher T, Neutze R et al. (2007). Light stimulates growth of proteorhodopsin-containing marine Flavobacteria. Nature 445: 210–213.
González JM, Kiene RP, Moran MA . (1999). Transformations of sulfur compounds by an abundant lineage of marine bacteria in the α-subclass of the class proteobacteria. Appl Environ Microbiol 65: 3810–3819.
Harada H, Rouse M-A, Sunda W, Kiene RP . (2004). Latitudinal and vertical distributions of particle-associated DMSP lyase activity in the western North Atlantic Ocean. Can J Fish Aquat Sci 61: 700–711.
Hewson I, Poretsky RS, Dyhrman ST, Zielinski B, White AE, Tripp HJ et al. (2009). Microbial community gene expression within colonies of the diazotroph, Trichodesmium, from the Southwest Pacific Ocean. ISME J 3: 1286–1300.
Howard EC, Henriksen JR, Buchan A, Reisch CR, Bürgmann H, Welsh R et al. (2006). Bacterial taxa that limit sulfur flux from the ocean. Science 314: 649–652.
Howard EC, Sun S, Biers EJ, Moran MA . (2008). Abundant and diverse bacteria involved in DMSP degradation in marine surface waters. Environ Microbiol 10: 2397–2410.
Huson DH, Auch AF, Qi J, Schuster SC . (2007). MEGAN analysis of metagenomic data. Genome Res 17: 377–386.
Ingraham JL, Maaloe O, Neidhardt F . (1983). Growth of the bacterial cell. Sinauer Associates: Sunderland, Massachusetts.
Kanehisa M, Goto S . (2000). KEGG: kyoto encyclopedia of genes and genomes. Nucl Acids Res 28: 27–30.
Keller MD, Bellows WK, Guillard RRL . (1989). Dimethyl sulfide production in marine phytoplankton. In: Saltzman E and Cooper WJ (eds). Biogenic Sulfur in the Environment. Am. Chemical Soc.: Washington, pp 167–182.
Kiene RP . (1990). Dimethyl sulfide production from dimethylsulfoniopropionate in coastal seawater samples and bacterial cultures. Appl Environ Microbiol 56: 3292–3297.
Kiene RP, Linn LJ . (2000a). Distribution and turnover of dissolved DMSP and its relationship with bacterial production in the Gulf of Mexico. Limnol Oceanogr 45: 849–861.
Kiene RP, Linn LJ . (2000b). The fate of dissolved dimethylsulfoniopropionate (DMSP) in seawater: tracer studies using 35S-DMSP. Geochim Cosmochim Acta 64: 2797–2810.
Kiene RP, Linn LJ, Bruton JA . (2000). New and important roles for DMSP in marine microbial communities. J Sea Res 43: 209–224.
Kiene RP, Linn LJ, González JM, Moran MA, Bruton JA . (1999). Dimethylsulfoniopropionate and methanethiol are important precursors of methionine and protein-sulfur in marine bacterioplankton. Appl Environ Microbiol 65: 4549–4558.
Kiene RP, Slezak D . (2006). Low dissolved DMSP concentrations in seawater revealed by small-volume gravity filtration and dialysis sampling. Limnol Oceanogr Meth 4: 80–95.
Ledyard KM, Dacey JWH . (1996). Microbial cycling of DMSP and DMS in coastal and oligotrophic seawater. Limnol Oceanogr 41: 33–40.
Li W, Godzik A . (2006). Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22: 1658–1659.
Lovelock JE, Maggs RJ, Rasmussen RA . (1972). Atmospheric dimethyl sulfide and the natural sulfur cycle. Nature 237: 452–453.
Malmstrom RR, Kiene RP, Cottrell M, Kirchman DL . (2004b). Contribution of SAR11 bacteria to C, N, and S cycling in the North Atlantic Ocean. Appl Environ Mircobiol 70: 4129–4135.
Malmstrom RR, Kiene RP, Kirchman DL . (2004a). Identification and enumeration of bacteria assimilating dimethylsulfoniopropionate (DMSP) in the North Atlantic and Gulf of Mexico. Limnol Oceanogr 49: 597–606.
Malmstrom RR, Kiene RP, Vila M, Kirchman DL . (2005). Dimethylsulfoniopropionate (DMSP) assimilation by Synechococcus in the Gulf of Mexico and Northwest Atlantic Ocean. Limnol Oceanogr 50: 1924–1931.
Massana R, Pedros-Alio C, Casamayor EO, Gasol JM . (2001). Changes in marine bacterioplankton phylogenetic composition during incubations designed to measure biogeochemically significant parameters. Limnol Oceanogr 46: 1181–1188.
McGillicuddy DJ . (1998). Influence of mesoscale eddies on new production in the Sargasso Sea. Nature 394: 263–266.
Moran MA . (2008). Marine prokaryotic genomics and metagenomics. In: Kirchman DL (ed). Microbial Ecology of the Oceans. Wiley: NY.
Moran MA . (2009). Crystal ball—2009. Environ Microbiol Rep 1: 3–26.
Moran MA, Buchan A, Gonzalez JM, Heidelberg JF, Whitman WB, Kiene RP et al. (2004). Genome sequence of Silicibacter pomeroyi reveals adaptations to the marine environment. Nature 432: 910–913.
Mou X, Sun S, Edwards RA, Hodson RE, Moran MA . (2008). Bacterial carbon processing by generalist species in the coastal ocean. Nature 451: 708–711.
Neufeld JD, Schäfer H, Cox MJ, Boden R, McDonald IR, Murrell JC . (2007). Stable-isotope probing implicates Methylophaga spp and novel Gammaproteobacteria in marine methanol and methylamine metabolism. ISME J 1: 480–491.
Poretsky RS, Bano N, Buchan A, LeCleir G, Kleikemper J, Pickering M et al. (2005). Analysis of microbial gene transcripts in environmental samples. Appl Environ Microbiol 71: 4121–4126.
Poretsky RS, Hewson I, Sun S, Allen AE, Zehr JP, Moran MA . (2009). Comparative day/night metatranscriptomic analysis of microbial communities in the North Pacific Subtropical Gyre. Environ Microbiol 11: 1358–1375.
Poretsky RS, Sun S, Mou X, Moran MA . (2010). Transporter genes expressed by coastal bacterioplankton in response to dissolved organic carbon. Environ Microbiol 12: 616–627.
Rodriguez-Brito B, Rohwer F, Edwards RA . (2006). An application of statistics to comparative metagenomics. BMC Bioinformatics 7: 162.
Seshadri R, Kravitz SA, Smarr L, Gilna P, Frazier M . (2007). CAMERA: A community resource for metagenomics. PLoS Biology 5: e75. doi:10.1371/journal.pbio.0050075.
Shi Y, Tyson GW, DeLong EF . (2009). Metatranscriptomics reveals unique microbial small RNAs in the ocean's water column. Nature 459: 266–269.
Simó R . (2001). Production of atmospheric sulfur by oceanic plankton: biogeochemical, ecological and evolutionary links. Trends Ecol Evol 16: 287–294.
Simó R, Archer SD, Pedrós-Alió C, Gilpin L, Stelfox-Widdicombe CE . (2002). Coupled dynamics of dimethylsulfoniopropionate and dimethylsulfide cycling and the microbial food web in surface waters of the North Atlantic. Limnol Oceanogr 47: 53–61.
Simó R, Vila-Costa M, Alonso-Saez L, Cardelus C, Guadayol O, Vázquez-Domínguez E et al. (2009). Annual DMSP contribution to S and C fluxes through phytoplankton and bacterioplankton in a NW Mediterranean coastal site. Aquat Microb Ecol 57: 43–55.
Sogin ML, Morrison HG, Huber JA, Welch DM, Huse SM, Neal PR et al. (2006). Microbial diversity in the deep sea and the underexplored ‘rare biosphere’. Proc Natl Acad Sci 103: 12115–12120.
Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV et al. (2003). The COG database: an updated version includes eukaryotes. BMC Bioinformatics 11: 41.
Todd JD, Curson ARJ, Dupont CL, Nicholson P, Johnston AWB . (2009). Corrigendum: The dddP gene, encoding a novel enzyme that converts dimethylsulfoniopropionate into dimethyl sulfide, is widespread in ocean metagenomes and marine bacteria and also occurs in some Ascomycete fungi. Environ Microbiol 11: 1624–1625.
Todd JD, Curson ARJ, Nikolaidou-Katsaraidou N, Brearley CA, Watmough NJ, Chan Y et al. (2010). Molecular dissection of bacterial acrylate catabolism—unexpected links with dimethylsulfoniopropionate catabolism and dimethyl sulfide production. Environ Microbiol 12: 327–343.
Todd JD, Rogers R, Li YG, Wexler M, Bond PL, Sun L et al. (2007). Structural and regulatory genes required to make the gas dimethyl sulfide in bacteria. Science 315: 666–669.
Tripp HJ, Kitner JB, Schwalbach MS, Dacey JWH, Wilhelm LJ, Giovannoni SJ . (2008). SAR11 marine bacteria require exogenous reduced sulphur for growth. Nature 452: 741–744.
Venter JC, Remington K, Heidelberg JF, Halpern AL, Rusch D, Eisen JA et al. (2004). Environmental Genome Shotgun Sequencing of the Sargasso Sea. Science 304: 66–74.
Vila M, Simó R, Kiene RP, Pinhassi J, Gonzalez JM, Moran MA et al. (2004). Use of microautoradiography combined with fluorescence in situ hybridization to determine dimethylsulfoniopropionate incroporation by marine bacterioplankton taxa. Appl Environ Microbiol 70: 4648–4657.
Vila-Costa M, Kiene RP, Simó R . (2008). Seasonal variability of the dynamics of dimethylated sulfur compounds in a coastal northwest Mediterranean site. Limnol Oceanogr 53: 198–211.
Vila-Costa M, Pinhassi J, Alonso C, Pernthaler J, Simó R . (2007). An annual cycle of dimethylsulfoniopropionate-sulfur and leucine assimilating bacterioplankton in the coastal NW Mediterranean. Environ Microbiol 9: 2451–2463.
Vila-Costa M, Simó R, Harada H, Gasol JM, Slezak D, Kiene RP . (2006). Dimethylsulfoniopropionate uptake by marine phytoplankton. Science 314: 652–654.
Yoch DC . (2002). Dimethylsulfoniopropionate: its sources, role in the marine food web, and biological degradation to dimethylsulfide. Appl Environ Microbiol 68: 5804–5815.
Zubkov MV, Fuchs BM, Archer SD, Kiene RP, Amann R, Burkill PH . (2002). Rapid turnover of dissolved DMS and DMSP by defined bacterioplankton communities in the stratified euphotic zone of the North Sea. Deep-Sea Res Part II 49: 3017–3038.
Acknowledgements
We thank J Dacey and D Toole for the invitation to join BATS cruises in the Sargasso Sea, N Levine and A Neely for their help on the cruises, and C Reisch for DMSP synthesis. The captain and crew of the R/V Atlantic Explorer, the BATS team, especially S Bell, are gratefully acknowledged. W Ye provided essential help in cruise arrangements, D del Valle and R Kiene conducted DMSP analyses, and O Garcia and C English provided graphics assistance. This study was funded by a Marie Curie Fellowship (to MVC), the Gordon and Betty Moore Foundation, and the National Science Foundation (OCE0724017 to MAM and OCE0425166 to J Dacey and D Toole).
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on The ISME Journal website
Rights and permissions
About this article
Cite this article
Vila-Costa, M., Rinta-Kanto, J., Sun, S. et al. Transcriptomic analysis of a marine bacterial community enriched with dimethylsulfoniopropionate. ISME J 4, 1410–1420 (2010). https://doi.org/10.1038/ismej.2010.62
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2010.62
Keywords
This article is cited by
-
The ecological responses of bacterioplankton during a Phaeocystis globosa bloom in Beibu Gulf, China highlighted by integrated metagenomics and metatranscriptomics
Marine Biology (2022)
-
Single-taxon field measurements of bacterial gene regulation controlling DMSP fate
The ISME Journal (2015)
-
Microbial controls on DMSP degradation and DMS formation in the Sargasso Sea
Biogeochemistry (2014)
-
Sizing up metatranscriptomics
The ISME Journal (2013)
-
The activity level of a microbial community function can be predicted from its metatranscriptome
The ISME Journal (2012)
