
Abstract
The spontaneous microaggregation of platelets (SMAPs) is a marker for the prognosis of patients with cardiovascular diseases. Coupling factor 6 (CF6) binds to the plasma membrane ATP synthase and functions as a pro-atherogenic molecule in the cardiovascular system. However, the role of CF6 in SMAPs and stroke remains unknown. In 650 consecutive patients, including those with acute-onset stroke, and 20 control subjects, platelet-rich plasma (PRP) was obtained, and SMAP was measured using a laser light-scattering aggregometer. The cytosolic cyclic adenosine monophosphate (cAMP) concentration in platelets was measured using an enzyme-linked immunosorbent assay. CF6 increased SMAPs in patients and control subjects to a similar degree by binding to the α- and β-subunits of ATP synthase and inducing intracellular acidosis. It was abolished by PRP pretreatment with antibodies against CF6, and the α- or β-subunit of the plasma membrane ATP synthase, and the ATP synthase inhibitor efrapeptin. CF6 increased SMAPs in patient groups with and without antiplatelet therapy to a similar degree, and no difference was found among the subgroups taking aspirin, thienopyridine or cilostazol. The cytosolic cAMP concentration in platelets was decreased by CF6 in the presence of the direct adenylate cyclase activator forskolin. Pretreatment of PRP with the Gs activator cholera toxin blocked the decrease, whereas the Gi inactivator pertussis toxin and cilostazol had no influence. The CF6-induced acceleration of SMAPs was suppressed by cholera toxin but not by cilostazol or pertussis toxin. CF6 enhanced SMAPs by decreasing cytosolic cAMP. Because it was observed irrespective of antiplatelet agents, CF6 appears to be a novel target for antiplatelet therapy.
Similar content being viewed by others


Introduction
Platelet activation and aggregation are fundamental processes in the development of atherosclerosis and thrombosis, both of which contribute to cardiovascular risk.1, 2 When activated, platelets aggregate and form microaggregates of a few cells as the initial response to various stimuli, and when exposed to prolonged stimulation, larger and tighter platelet aggregates appear.3 Spontaneous microaggregation of platelets (SMAPs), which is composed of small-sized platelet aggregates, occurs with stirring force only and has no requirement for exogenous agonist stimulation. The production of small-sized platelet aggregates is important in initial thrombus formation,4 and SMAP is a marker for the prognosis of patients with cardiovascular diseases such as myocardial infarction.5 However, the occurrence of SMAPs in stroke remains unclear. The laser light-scattering method enabled us to detect small-sized platelet aggregates.
The presence of the molecular rotary motor F1Fo complex ATP synthase on the plasma membrane has been reported in several types of cells, and its novel functions have been disclosed.6, 7, 8, 9 In hepatocytes, F1Fo complex simulation by apoprotein A-I triggers the endocytosis of holo-high-density lipoprotein particles by a mechanism that depends on the generation of ADP.7 In endothelial cells, the interaction of the β-subunit of the F1 motor with angiostatin attenuates ATP generation and angiogenesis.8, 9 The ecto-F1Fo complex acts as the receptor for lipid uptake and angiogenesis, but its ligands and functions are not fully understood. We recently identified a circulating peptide, coupling factor 6 (CF6), as a novel ligand for the ecto-F1Fo complex in vascular endothelial and smooth muscle cells. CF6 is composed of 76 amino acids and is released from vascular endothelial cells by shear stress and tumor necrosis factor-α.10, 11 After binding to protrusive F1, CF6 forces the backward rotation of Fo, thereby acting as a stimulator of the proton-importing transporter at the plasma membrane, and functions as a pro-atherogenic molecule in the cardiovascular system.6 CF6 attenuates both prostacyclin generation via the inhibition of cytosolic phospholipase A2 and nitric oxide generation via the upregulation of asymmetric dimethylarginine or the inhibition of endothelial nitric oxide synthase phosphorylation in vascular endothelial cells.12, 13, 14 CF6 elevates arterial blood pressure in spontaneously hypertensive rats and enhances angiotensin II-induced vasoconstriction in resistance arterioles.15, 16 The plasma concentration of CF6 in patients with essential hypertension is elevated compared with that in normotensive subjects, and the change in blood pressure because of high salt intake is positively correlated with the change in the plasma concentration of CF6.17 In CF6-overexpressing transgenic mice, a high salt intake induces cardiac systolic dysfunction and nicotinamide adenine dinucleotide phosphate oxidase upregulation18 and salt-sensitive hypertension by Rac1 activation.19 A high-sucrose diet induces type 2 diabetes by inactivating the insulin receptor,20 and exercise-induced physiological cardiac hypertrophy is attenuated by downregulating AKt signaling.21
Although the role of CF6 in the cardiovascular system has been uncovered, the effect of CF6 on SMAPs still remains unknown. In the present study, we investigated SMAPs in patients with stroke and the role of CF6 this process.
Methods
Study patients
The protocol of the present study was approved by the ethics committee of the Hirosaki Stroke and Rehabilitation Center, and informed consent was obtained from the patients or their family (Table 1). A total of 650 consecutive patients who were transferred to the emergency room of the Hirosaki Stroke and Rehabilitation Center for the suspicion of acute-onset stroke during the 6-month period from August 2010 through January 2011 and underwent head CT and MRI were enrolled. Twenty age- and sex-matched volunteers were also enrolled. Peripheral venous blood was sampled to measure CF6 and biochemical parameters. PRP was obtained from 424 patients with a recent onset of stroke (atherothrombotic stroke, 173; lacunar, 71; cardioembolic, 95; hemorrhagic, 85) and 226 non-stroke patients upon arrival at the emergency room in addition to 20 control subjects. The non-stroke patients suffered from headache, vertigo and numbness, but acute-onset stroke was excluded by neurological findings and imaging studies. The diagnosis of non-stroke patients included disturbance of consciousness by infection, drug intoxication and hypoglycemia, brain tumor, peripheral vertigo, peripheral neuropathy and epilepsy. Some patients had a past history of cardiovascular disease, including cerebral infarction and myocardial infarction. The control subjects did not have a past history of cardiovascular disease, but in some cases, antiplatelet medicines were administered to prevent stroke or peripheral artery disease due to risk factors such as hypertension, dyslipidemia and diabetes.
Table 1 shows the clinical profiles for stroke and non-stroke patients, and control subjects. Age and gender did not differ among the three groups. Compared with control subjects, the incidence of cardiovascular risk factors including hypertension, dyslipidemia, diabetes and smoking was higher in patients with stroke. In patients without stroke, the risk factors were similar to the control subjects. The patients were administered aspirin (100 mg per day, n=99), thienopyridine (clopidogrel at 75–150 mg per day or ticlopidine at 100–200 mg per day, n=76) and cilostazol(100–200 mg per day, n=55).
Detection of platelet microaggregation
The light intensities of small- (9–25 μm), medium- (25–50 μm) and large- (50–70 μm) sized platelet aggregates were measured using a laser light-scattering aggregometer (model PA-200C; Kowa, Tokyo, Japan) with or without CF6. Briefly, PRP was obtained from blood collected into sodium citrate (14 μmol l−1) by immediate centrifugation at 155 × g for 12 min at room temperature. Platelet microaggregation was determined by measuring the light-scattering intensity on a PA-200 aggregometer. SMAP was observed under low shear stress conditions at a stirring speed of 1000 r.p.m. (26 dyn cm−2) without stimulation by an exogenous agonist. This degree of low shear stress is considered to be the same degree of shear stress occurring in the arterial stream, suggesting that this system provides a condition that mimics the state of platelets in the artery. In some cases, ADP-induced platelet aggregation was measured. The data were recorded as a two-dimensional graph showing the change in total light intensity over time, expressed as a cumulative summation at 10-s intervals of scattered light intensity and the number of particles corresponding to that intensity in terms of particle size (volts × counts per second). Particles with an intensity of 25–400 mV represented small aggregates (9–25 μm). The degrees of SMAP- and ADP-induced platelet aggregation were described by measuring the area under the curve (AUC) of each detection line for 10 min. AUC data were expressed as × 105 particles.
Flow cytometry analysis
Cell surface P-selectin and the α- and β-subunits of ATP synthase were analyzed using a FACScan.10 After platelets were incubated with or without CF6 at 10−7 M for 30 min, they were washed and reacted with saturating concentrations of anti-P-selectin and anti-α- or anti-β-subunit of ATP synthase antibodies (all diluted 1:1000) and isotype control antibodies in phosphate-buffered saline containing 1% bovine serum albumin for 30 min at room temperature. After washing three times with phosphate-buffered saline, they were stained with fluorescein isothiocyanate-conjugated goat anti-rabbit or anti-mouse IgG for 30 min and then analyzed using a FACScan (BD FACSCanto II, Franklin Lakes, NJ, USA).
Western blot analysis
The α- and β-subunits of ATP synthase were measured by a western blot analysis as previously described.6 The plasma membrane was obtained by a centrifugation strategy. The lysate of the plasma membrane was subjected to SDS-polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride membrane (Bio-Rad Laboratories, Hercules, CA, USA). After blocking for 1 h, the membranes were incubated with primary antibody for the α- or β-subunit of ATP synthase, followed by the secondary antibody. The protein bands were detected using the ECL Plus detection system (Amersham Pharmacia Biotech, Piscataway, NJ, USA). A densitometric analysis was performed with NIH imaging software.
Intracellular pH measurement
Intracellular pH was measured using BCECF-AM as previously reported.6 Platelets were incubated in Hank’s balanced salt solution containing BCECF-AM at 4 μg ml−1 for 20 min at 37 °C. Excess dye was removed by centrifugation, and the cells were suspended in nominally HCO3−-free Krebs–Henseleit buffered with 20 mmol l−1 Hepes containing 0.2% fatty acid-free BSA. The suspension was transferred to a cuvette that was placed in the temperature-controlled chamber of a Shimadzu 5000 luminescence spectrophotometer (Shimadzu, Kyoto, Japan), maintained at 37 °C. Using a dual excitation fast-filter accessory, the sample was excited at 495 nm and 440 nm successively, and the fluorescence emission was measured at 535 nm. The ratio of the fluorescence intensity measured using the two excitation wavelengths (495 nm/440 nm) provides a quantitative measure of pH.
Radioimmunoassay for CF6
CF6 was measured by radioimmunoassay as previously reported.10 Briefly, Sep-Pak C18 cartridges were loaded with plasma acidified with HCl. After washing with water, the absorbed materials were eluted with 60% acetonitrile containing 0.1% trifluoroacetic acid and submitted to radioimmunoassay. The CF6 standard and the unknown samples were incubated with anti-CF6 antiserum diluent (1:1200) for 12 h, and then the tracer solution (18 000–20 000 c.p.m.) was added. After incubation for 24 h, anti-rabbit IgG goat serum diluent containing 10% polyethylene glycol 6000 and rabbit IgG were added, and the radioactivity of the precipitate was measured in a γ-counter.
Determination of cyclic AMP
Cyclic adenosine monophosphate (cAMP) was measured using an enzyme-linked immunosorbent assay (ELISA). After the incubation of PRP with the direct adenylate cyclase activator forskolin at 10−5 M and/or cilostazol at 10−5 M for 15 min at room temperature, CF6 was added to the mixture at 10−6 M and was incubated for 15 min at room temperature. In some experiments, platelets were pretreated with cholera toxin and/or pertussis toxin for 3 h. The reaction was stopped by cooling and centrifugation at 2000 × g for 5 min. Platelets were washed with phosphate-buffered saline and then dissolved in 0.1 N HCl. After centrifugation at 2000 × g for 5 min at 4 °C, cAMP was measured in the supernatant using an ELISA kit (Cayman Chemical Company, Ann Arbor, MI, USA).
Statistical analysis
All results are expressed as the mean±s.e.m. Differences between groups were examined for statistical significance using Student’s t test, the Wilcoxon paired test and the Kruskal–Wallis analysis of variance followed by Sheffé’s test. Relationships between two continuous variables were assessed by a regression analysis using the Pearson correlation coefficient. Differences in items were analyzed by the χ2-test or Fisher’s exact probability test. The level of significance was less than 0.05.
Results
Interaction between CF6 and human platelets
As shown in Figure 1A, fluorescence-assisted flow cytometry confirmed the cell-surface localization of the α- and β-subunits in platelets. The selectivity of the response to the α- and β-subunit antibodies was confirmed by the low fluorescein isothiocyanate signal obtained with the IgG isotype control. After preincubating the cells with an excess of free CF6, the immunoreactivity for the α- and β-subunit antibodies was decreased at the platelet surface, suggesting the cell surface interaction of free CF6 with the α-and β-subunits of ATP synthase. Identical results were observed in three separate experiments using different platelet samples. As shown in Figure 1B, the presence of the α- and β-subunits on the platelet surface was directly confirmed by western blot analysis. Figure 1C illustrates representative tracings of the intracellular pH after CF6 administration to platelets. CF6 at 10−7 M decreased intracellular pH within seconds, gradually reaching the maximal decrease and returning to baseline after several minutes. Pretreatment with efrapeptin at 10−5 M abolished the reduction of intracellular pH induced by CF6 at 10−7 M. Amiloride at 10−3 M, an inhibitor of sodium–hydrogen exchange, promptly and remarkably decreased intracellular pH, suggesting that sodium–hydrogen exchange is intact after treatment with efrapeptin at 10−5 M and CF6 at 10−7 M.

(A) Representative tracings of flow cytometry for platelet surface associated α- and β-subunits of ATP synthase before and after preincubating with CF6 at 10-7 M. (B) The α- and β-subunits of ATP synthase were measured by western blot analysis. GAPDH; glyceraldehyde-3-phosphate dehydrogenase. (C) Effect of CF6 on intracellular pH. (a) Intracellular pH after administration of CF6 at 10-7 M. (b) Effect of pretreatment with efrapeptin (EF) at 10-5 M on intracellular pH in response to CF6 at 10-7 M.
Effect of CF6 on SMAPs in patients and control subjects
Figure 2B shows representative waveforms of SMAPs in the presence or absence of CF6 at 10−7 M in a patient with atherothrombotic stroke and a control subject. The light intensity of small-sized platelet aggregates gradually increased regardless of the presence of CF6 even though the optical density was unchanged. The AUC of small-sized aggregates in spontaneous platelet aggregation (mV × count × min) was greater in patients with stroke than in control subjects (1.34±0.08 × 107 vs. 0.80±0.18 × 107, P<0.01). As shown in Figure 2A (white column), the AUCs were similar among the types of stroke (atherothrombotic stroke, 1.28±0.16 × 107; lacunar, 0.95±0.15 × 107; cardioembolic, 1.28±0.21 × 107; and hemorrhagic, 1.44±0.23 × 107). The AUC in patients without stroke (1.47±0.15 × 107) was similar to that in patients with stroke. Among patients with and without stroke, SMAPs did not differ in the presence or absence of risk factors, including diabetes. The plasma level of CF6 was higher in patients with and without stroke than in control subjects (16.6±0.2 and 15.8±0.3 ng ml−1 vs. 10.1±0.4 ng ml−1, both P<0.05) and was higher in patients with stroke than in those without stroke (P<0.05). Plasma CF6 was not correlated with age, but there was a weak positive correlation between plasma CF6 and SMAPs after adjusting for risk factors (r=0.138, P<0.05). Even in patients without antiplatelet medication, the CF6 concentration was positively but weakly correlated with SMAP formation (r=0.137, P<0.05).

(A) AUC of small-sized aggregation in spontaneous platelet aggregation in the presence or absence of CF6. (B) Representative waveforms of spontaneous platelet aggregation in the presence or absence of CF6 at 10-7 M in patient with atherothrombotic stroke and control subject. (Ca) Representative tracings of flow cytometry for platelet surface associated P-selectin in stroke patient and control subject. (Cb) Fluorescein isothiocyanate-A (FITC-A) mean in stroke patients and control subjects, n=6. (Cc) Representative tracings of flow cytometry for platelet surface associated P-selectin in the presence or absence of CF6 at 10-7 M in stroke patient. (Cd) Change in FITC-A mean in the presence or absence of CF6 at 10-7 M in stroke patients, n=12.
CF6 administration at 10−7 M increased the AUC of small-sized aggregates to a similar degree in patients with and without stroke and control subjects (Figure 2A, black column). Baseline platelet activation markers were evaluated, and surface P-selectin was increased in patients with stroke compared with the control subjects (P<0.05) (Figure 2C top) and was increased 6.3±2.5% by CF6 at 10−7 M (P<0.05) (Figure 2C, bottom). CF6 at 10−7 M increased the AUC of large-sized aggregates stimulated with 2 μM ADP from 3.08±0.20 × 107 to 3.52±0.20 × 107 (P<0.05) in patients with atherothrombotic stroke.
We further compared the levels of SMAPs at baseline or those after CF6 addition between the groups treated with and without antiplatelet agents. Figure 3a shows representative waveforms of SMAPs in the presence or absence of CF6 at 10−7 M in patients taking aspirin or clopidogrel. The light intensity of small-sized platelet aggregates gradually increased irrespective of CF6, and it was greater in the presence of CF6 than in the absence of CF6. As shown in Figure 3b, CF6 at 10−7 M increased SMAPs to a similar degree in the patient groups with and without antiplatelet agents. There was no difference among the subgroups of the patients taking aspirin, thienopyridine and cilostazol. CF6 receptor binding was inhibited to evaluate the role of CF6 in SMAPs. Pretreatment of PRP with antibodies against CF6 and the α- or β-subunit of ecto-F1 and the F1 inhibitor efrapeptin blocked the CF6-induced increase in SMAPs (Figure 4).

(a) Representative waveforms of spontaneous platelet aggregation in the presence or absence of CF6 at 10-7 M in patient with aspirin and clopidogrel. (b) AUC of small-sized aggregation in spontaneous platelet aggregation in the presence or absence of CF6.

AUC of small-sized aggregation in spontaneous platelet aggregation in the presence or absence of CF6. Pretreatment of the platelet-rich plasma with antibodies against CF6 (a), α-subunit (b), β-subunit (c) and F1 inhibitor efrapeptin (d), all n=10.
Role of Gs and Gi in CF6-mediated SMAPs and cAMP in platelets
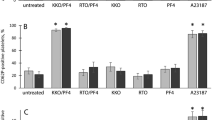
As shown in Figure 5a, cytosolic cAMP was not affected at baseline by cilostazol, pertussis toxin or cholera toxin. Treatment with CF6 at 10−6 M for 15 min decreased cytosolic cAMP (Figure 5b). Cilostazol treatment of platelets did not influence the decrease in cytosolic cAMP, but it was abolished by cholera toxin. The addition of pertussis toxin did not affect the decrease in cytosolic cAMP induced by CF6 at 10−6 M. Figures 5c and d show SMAPs in the presence or absence of CF6 at 10−7 M after the addition of cilostazol, pertussis toxin and cholera toxin. The AUC of small-sized aggregates in SMAPs was decreased by cilostazol but was not influenced by pertussis toxin or cholera toxin (Figure 5c). The CF6-induced increase in SMAPs was suppressed by cholera toxin but not by cilostazol or pertussis toxin (Figure 5d).

(a) Cytosolic cAMP concentration at baseline after addition of cilostazol, pertussis toxin and cholera toxin. (b) Change in cytosolic cAMP by addition of CF6 in PRP pretreated with cilostazol, pertussis toxin and cholera toxin, n=6. (c) AUC of small-sized aggregates in spontaneous platelet aggregation in after addition of cilostazol, pertussis toxin and cholera toxin. (d) Change in AUC of small-sized aggregates by the addition of CF6 in PRP pretreatment with cilostazol, pertussis toxin and cholera toxin, n=3.
Discussion
The major findings of this study were as follows. CF6 increased SMAPs by binding to the α- and β-subunits of ATP synthase, subsequently leading to intracellular acidosis. This CF6-induced increase in SMAPs was abolished by PRP pretreatment with antibodies against CF6 and the α- or β-subunit of the plasma membrane ATP synthase and the F1 inhibitor efrapeptin. Compared with control subjects, SMAP was increased in patients with and without stroke. CF6 increased SMAPs in the patient groups with and without antiplatelet therapy to a similar degree, and no difference in SMAPs was found among the subgroups taking aspirin, thienopyridine and cilostazol. Platelet cytosolic cAMP was decreased by CF6 in the presence of forskolin. PRP pretreatment with cholera toxin blocked the decrease, but neither pertussis toxin nor cilostazol had an effect on the cAMP decrease. The CF6-induced acceleration of SMAPs was suppressed by cholera toxin but not by cilostazol or pertussis toxin. This is the first report demonstrating that CF6 enhances SMAPs in patients with and without stroke and control subjects by decreasing cytosolic cAMP through Gs signaling regardless of antiplatelet therapy.
Effect of CF6 on SMAPs
CF6 is a component of the mitochondrial ATP synthase,22, 23 but circulating CF6 was recently shown to exert profound effects on the cardiovascular system.6, 12, 13, 14, 15, 16 CF6 binds to F1 of the plasma membrane ATP synthase in vascular endothelial and smooth muscle cells, and forces the backward rotation of Fo, resulting in intracellular acidosis.6 We first investigated the direct interaction of CF6 with the plasma membrane ATP synthase at the surface of human platelets using flow cytometry. The results showed that platelet incubation with an excess of free CF6 reduced the immunoreactivity for the α- and β-subunit antibodies, suggesting that CF6 interacted with the plasma membrane ATP synthase at the platelet surface. We further showed that CF6 decreased the intracellular pH within seconds and continued for several minutes, suggesting that CF6 functioned as a bioactive agent in the platelets as previously described in the cardiovascular system.
Because platelet microaggregates are produced in the early phase of platelet activation and are considered to potentially aggravate thrombus formation, the detection of platelet microaggregation seems important for the primary and secondary prevention of cardiovascular events. SMAP is observed under a low shear stress that mimics the state of the arterial blood stream. SMAPs at baseline in this study was enhanced in patients compared with control subjects, as described previously.24 We investigated the effect of CF6 on SMAPs and clearly showed that CF6 at 10−7 M increased SMAPs in patients with and without stroke and control subjects to a similar degree. Like other vasoactive substances, the plasma level of CF6 (10−9–10−8 M) was lower than the critical concentration (10−7 M) at which SMAPs was increased in vitro. However, we have previously shown that the intravenous administration of anti-CF6 antibody to rats counteracts its biological effects.15 Thus, the lower concentration of CF6 also appears to be active in vivo. According to the interaction study, it is likely that CF6 enhances SMAPs after binding to the α-and β-subunits of ATP synthase and inducing intracellular acidosis. This was confirmed by the present observation that PRP pretreatment with antibodies against CF6 and the α- or β-subunit of the plasma membrane ATP synthase and its inhibitor efrapeptin blocked the CF6-induced enhancement of SMAPs.
Many of the included patients were administered antiplatelet agents as monotherapy or in a combination therapy. However, it should be emphasized that CF6-induced SMAP formation was similar between patient groups with and without antiplatelet therapy. Moreover, there was no difference among the subgroups taking aspirin, thienopyridine and cilostazol. The prophylactic use of antiplatelet agents such as low-dose aspirin is widely recommended, but aspirin alone is insufficient to prevent thrombosis in diabetic patients with angiopathy.25 The clinical results shown by the combination therapy in the MATCH and CHARISMA trials also failed to demonstrate a convincing benefit compared with antiplatelet monotherapy for the secondary prevention of major cardiovascular events.26, 27 Antiplatelet therapy did not suppress the CF6-induced enhancement of SMAPs. Thus, CF6 inhibition may be important for the primary and secondary prevention of cardiovascular events.
Mechanism for the increase in SMAPs by CF6
The secretion of ADP from platelet-dense granules is closely associated with SMAPs by transforming resting platelets into activated platelets.28, 29 ADP interacts with its G-protein-coupled receptors at the platelet surface, namely P2Y1 coupled to Gq and P2Y12 coupled to Gi, and amplifies the platelet activation response.30, 31 Of the two signaling pathways, ADP-mediated P2Y12 signaling has an important role in platelet activation induced by a decrease in cytosolic cAMP.31 In the present study, CF6 increased SMAPs even in patients who were administered the P2Y12 blocker thienopyridine to a similar degree to those who were not, suggesting that ADP-mediated P2Y12 signaling is not related to the action of CF6 and other mechanisms might be involved. It is noteworthy that both the Gs-coupled receptor and the cAMP destruction enzyme are associated with SMAPs via modulating the cytosolic concentration of cAMP and interfering with Ca2+ homeostasis, degranulation and integrin activation.32 With regard to the destruction enzyme, platelets express two cAMP phosphodiesterase isoforms, cGMP-stimulated PDE2 and cGMP-inhibited PDE3A. PDE3A is the most abundant isoform in platelets and has a 250-fold lower Km for cAMP than PDE2.33 The PDE3A inhibitors cilostamide and milrinone have been reported to raise basal cAMP levels and strongly inhibit thrombin-induced platelet activation.34 In the present study, CF6 increased SMAPs in patients administered the PDE3A inhibitor cilostazol to a similar degree as in those who were not. Taken together, these suggest that neither the Gi-coupled ADP receptor P2Y12 nor cGMP-inhibited PDE3A is related to the effect of CF6 but rather that Gs signaling might be involved.
To clarify the involvement of Gs in the mechanism by which CF6 increased SMAPs, we estimated the effect of the Gs activator cholera toxin on cAMP in the presence of CF6. CF6 decreased cytosolic cAMP in forskolin-treated platelets, suggesting that cAMP might have an important role in the CF6-induced acceleration of SMAPs. The decrease was abolished by PRP pretreatment with cholera toxin, whereas neither the Gi inactivator pertussis toxin nor cilostazol affected it. Additionally, the CF6-induced acceleration of SMAPs was suppressed by cholera toxin but not by cilostazol or pertussis toxin. Overall, these observations suggest that Gs signaling is associated with a CF6-induced decrease in cAMP in human platelets and might be involved in accelerated SMAPs following CF6 administration.
Clinical implications
In this study, SMAP was greater at baseline in patients compared with control subjects, as described previously.24 The greater SMAPs may be associated with acute-onset stroke and a past history of cardiovascular diseases. Because SMAP is a marker for the prognosis of patients with atherosclerosis, increased SMAPs and a further increase in SMAPs induced by CF6 may be involved in the onset and progression of cardiovascular events.
The plasma level of CF6 is elevated in a number of cardiovascular disorders. We and others have shown that circulating CF6 is elevated in patients with hypertension, acute myocardial infarction, end-stage renal disease, stroke and diabetes,17, 35, 36, 37, 38 all of which predispose patients to the development of atherosclerosis. Given the present finding and the widespread biological actions of CF6 described previously, including the inhibition of prostacyclin and nitric oxide, CF6 appears to be a pro-atherogenic molecule in humans.12, 13, 14, 15, 16 To date, we have reported that salt restriction, vitamin C and vitamin B12 plus folic acid lower the plasma level of CF6 in patients with hypertension and stroke, and that peroxisomal proliferator-activated receptor γ ligand attenuates CF6 release from cultured vascular endothelial cells.17, 36, 39 Despite the absence of a specific inhibitor for CF6, lowering the circulating level of CF6 appears to effectively attenuate SMAPs.
Study limitations
The present study had some limitations. It was not possible to ascertain whether the activated platelet profile was a cause or consequence of the onset of stroke because of the nature of the cross-sectional study. The present study also could not address whether the incidence of future cardiovascular disease is higher in patients with SMAPs and whether the inhibition of the CF6-induced acceleration of SMAPs would be sufficient to prevent the development of cardiovascular disease.
In summary, the majority of patients showed an altered profile of platelet activation including SMAP events. Given the growing concern over cardiovascular consequences in patients, further follow-up and intervention studies are needed to establish whether CF6-induced SMAPs is involved in cardiovascular events and is a therapeutic target for their prevention.
References
Colwell JA, Nesto RW . The platelet in diabetes: focus on prevention of ischemic events. Diabetes Care 2003; 26: 2181–2188.
Ferroni P, Basili S, Falco A, Davì G . Platelet activation in type 2 diabetes mellitus. J Thromb Haemost 2004; 2: 1282–1291.
Ruggeri ZM . Platelets in atherothrombosis. Nat Med 2002; 8: 1227–1234.
Satoh K, Ozaki Y, Qi R, Yang L, Asazuma N, Yatomi Y, Kume S . Factors that affect the size of platelet aggregates in epinephrine-induced activation: a study using the particle counting method based upon light scattering. Thromb Res 1996; 81: 515–523.
Trip MD, Cats VM, van Capelle FJ, Vreeken J . Platelet hyperreactivity and prognosis in survivors of myocardial infarction. N Engl J Med 1990; 322: 1549–1554.
Osanai T, Magota K, Tanaka M, Shimada M, Murakami R, Sasaki S, Tomita H, Maeda N, Okumura K . Intracellular signaling for vasoconstrictor coupling factor 6: novel function of β-subunit of ATP synthase as receptor. Hypertension 2005; 46: 1140–1146.
Martinez LO, Jacquet S, Esteve JP, Rolland C, Cabezon E, Champagne E, Pineau T, Georgeaud V, Walker JE, Terce F, Collet X, Perret B, Barbaras R . Ectopic β-chain of ATP synthase is an apolopoprotein A-I receptor in hepatic HDL endocytosis. Nature 2003; 421: 75–79.
Moser TL, Stack MS, Asplin I, Enghild JJ, Hojrup P, Everitt L, Hubchak S, Schnaper HW, Pizzo SV . Angiostatin binds ATP synthase on the surface of human endothelial cells. Proc Natl Acad Sci USA 1999; 96: 2811–2816.
Moser TL, Kenan DJ, Ashley TA, Roy JA, Goodman MD, Misra UK, Cheek DJ, Pizzo SV . Endothelial cell surface F1-Fo ATP synthase is active in ATP synthesis and is inhibited by angiostatin. Proc Natl Acad Sci USA 2001; 98: 6656–6661.
Osanai T, Okada S, Sirato K, Nakano T, Saitoh M, Magota K, Okumura K . Mitochondrial coupling factor 6 is present on the surface of human vascular endothelial cells and released by shear stress. Circulation 2001; 104: 3132–3136.
Sasaki S, Osanai T, Tomita H, Matsunaga T, Magota K, Okumura K . Tumor necrosis factor α as an endogenous stimulator for circulating coupling factor 6. Cardiovasc Res 2004; 62: 578–586.
Osanai T, Kamada T, Fujiwara N, Katoh T, Takahashi K, Kimura M, Satoh K, Magota K, Kodama S, Tanaka T, Okumura K . A novel inhibitory effect on prostacyclin synthesis of coupling factor 6 extracted from the heart of spontaneously hypertensive rats. J Biol Chem 1998; 273: 31778–31783.
Tanaka M, Osanai T, Murakami R, Sasaki S, Tomita H, Maeda N, Satoh K, Magota K, Okumura K . Effect of vasoconstrictor coupling factor 6 on gene expression profile in human vascular endothelial cells: enhanced release of asymmetric dimethylarginine. J Hypertens 2006; 24: 489–497.
Kumagai A, Osanai T, Katoh C, Tanaka M, Tomita H, Morimoto T, Murakami R, Magota K, Okumura K . Coupling factor 6 downregulates platelet endothelial cell adhesion molecule-1 via c-Src activation and acts as a proatherogenic molecule. Atherosclerosis 2008; 200: 45–50.
Osanai T, Tanaka M, Kamada T, Nakano T, Takahashi K, Okada S, Sirato K, Magota K, Kodama S, Okumura K . Mitochondrial coupling factor 6 as a novel endogenous vasoconstrictor. J Clin Invest 2001; 108: 1023–1030.
Osanai T, Tomita H, Kushibiki M, Yamada M, Tanaka M, Ashitate T, Echizen T, Katoh C, Magota K, Okumura K . Coupling factor 6 enhances Src-mediated responsiveness to angiotensin II in resistance arterioles and cells. Cardiovasc Res 2009; 81: 780–787.
Osanai T, Sasaki S, Kamada T, Fujiwara N, Nakano T, Tomita H, Matsunaga T, Magota K, Okumura K . Circulating coupling factor 6 in human hypertension: role of reactive oxygen species. J Hypertens 2003; 21: 2323–2328.
Ashitate T, Osanai T, Tanaka M, Magota K, Echizen T, Izumiyama K, Yokoyama H, Shibutani S, Hanada K, Tomita H, Okumura K . Overexpression of coupling factor 6 causes cardiac dysfunction under high salt diet in mice. J Hypertens 2010; 28: 2243–2251.
Izumiyama K, Osanai T, Sagara S, Yamamoto Y, Itoh T, Sukekawa T, Nishizaki F, Magota K, Okumura K . Estrogen attenuates coupling factor 6-induced salt-sensitive hypertension and cardiac systolic dysfunction in mice. Hypertens Res 2012; 35: 539–546.
Osanai T, Tanaka M, Magota K, Tomita H, Okumura K . Coupling factor 6-induced activation of ecto-F1Fo complex induces insulin resistance, mild glucose intolerance and elevated blood pressure in mice. Diabetologia 2012; 55: 520–529.
Sagara S, Osanai T, Itoh T, Izumiyama K, Shibutani S, Hanada K, Yokoyama H, Yamamoto Y, Yokota T, Tomita H, Magota K, Okumura K . Overexpression of coupling factor 6 attenuates exercise-induced physiological cardiac hypertrophy by inhibiting PI3K/Akt signaling in mice. J Hypertens 2012; 30: 778–786.
Boyer PD . The binding change mechanism for ATP synthase: some probabilities and possibilities. Biochim Biophys Acta 1993; 1140: 215–250.
Walker JE, Fearnley IM, Gay NJ, Gibson BW, Northrop FD, Powell SJ, Runswick MJ, Saraste M, Tybulewicz VL . Primary structure and subunit stoichiometry of F1-ATPase from bovine mitochondria. J Mol Biol 1985; 184: 677–701.
Araki S, Matsuno H, Haneda M, Kyoya D, Kanno Y, Itho J, Kishi A, Isshiki K, Sugimoto T, Maegawa H, Kashiwagi A, Uzu T . Correlation between albuminuria and spontaneous platelet microaggregate formation in type 2 diabetic patients. Diabetes Care 2009; 32: 2062–2067.
Di Minno G, Violi F . Aspirin resistance and diabetic angiopathy: back to the future. Thromb Res 2004; 113: 97–99.
Diener HC, Bogousslavsky J, Brass LM, Cimminiello C, Csiba L, Kaste M, Leys D, Matias-Guiu J, Rupprecht HJ, MATCH investigators. Aspirin and clopidogrel compared with clopidogrel alone after recent ischaemic stroke or transient ischaemic attack in high-risk patients (MATCH): a randomised, double-blind, placebo-controlled trial. Lancet 2004; 364: 331–337.
Bhatt DL, Topol EJ . Clopidogrel added to aspirin versus aspirin alone in secondary prevention and high-risk primary prevention: rationale and design of the Clopidogrel for High Atherothrombotic Risk and Ischemic Stabilization, Management, and Avoidance (CHARISMA) trial. Am Heart J 2004; 148: 263–268.
Charo IF, Feinman RD, Detwiler TC . Interrelations of platelet aggregation and secretion. J Clin Invest 1977; 60: 866–873.
Coller BS . Platelet and thrombolytic therapy. N Engl J Med 1990; 322: 33–42.
Daniel JL, Dangelmaier C, Jin J, Ashby B, Smith JB, Kunapuli SP . Molecular basis for ADP-induced platelet activation I: evidence for three distinct ADP receptors on platelets. J Biol Chem 1998; 273: 2024–2029.
Foster CJ, Prosser DM, Agans JM, Zhai Y, Smith MD, Lachowicz JE, Zhang FL, Gustafson E, Monsma FJ, Wiekowski MT, Abbondanzo SJ, Cook DN, Bayne ML, Lira SA, Chintala MS . Molecular identification and characterization of the platelet ADP receptor targeted by thienopyridine antithrombotic drugs. J Clin Invest 2001; 107: 1591–1598.
Storey RF, Sanderson HM, White AE, May JA, Cameron KE, Heptinstall S . The central role of the P2T receptor in amplification of human platelet activation, aggregation, secretion and procoagulant activity. Br J Haematol 2000; 110: 925–934.
Schwarz UR, Walter U, Eigenthaler M . Taming platelets with cyclic nucleotides. Biochem Pharmacol 2001; 62: 1153–1161.
Manns JM, Brenna KJ, Colman RW, Sheth SB . Differential regulation of human platelet responses by cGMP inhibited and stimulated cAMP phosphodiesterases. Thromb Haemost 2002; 87: 873–879.
Osanai T, Nakamura M, Sasaki S, Tomita H, Saitoh M, Osawa H, Yamabe H, Murakami S, Magota K, Okumura K . Plasma concentration of coupling factor 6 and cardiovascular events in patients with end-stage renal disease. Kidney Int 2003; 64: 2291–2297.
Osanai T, Fujiwara N, Sasaki S, Metoki N, Saitoh G, Tomita H, Nishimura T, Shibitani S, Yokoyama H, Konta Y, Magota K, Okumura K . Novel pro-atherogenic molecule coupling factor 6 is elevated in patients with stroke: a possible linkage to homocysteine. Ann Med 2010; 42: 79–86.
Ding WH, Chu SY, Jiang HF, Cai DY, Pang YZ, Tang CS, Qi YF . Plasma mitochondrial coupling factor 6 in patients with acute myocardial infarction. Hypertens Res 2004; 27: 717–722.
Li XL, Xing QC, Gao YY, Dong B, Pang YZ, Jiang HF, Tang CS . Plasma level of mitochondrial coupling factor 6 increases in patients with type 2 diabetes mellitus. Int J Cardiol 2007; 117: 411–412.
Tomita H, Osanai T, Toki T, Sasaki S, Maeda N, Murakami R, Magota K, Yasujima M, Okumura K . Troglitazone and 15-deoxy-deta 12, 14-prostaglandin J2 inhibit shear-induced coupling factor 6 release in endothelial cells. Cardiovasc Res 2005; 67: 134–141.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Sukekawa, T., Osanai, T., Nishizaki, F. et al. Coupling factor 6 enhances the spontaneous microaggregation of platelets by decreasing cytosolic cAMP irrespective of antiplatelet therapy. Hypertens Res 36, 520–527 (2013). https://doi.org/10.1038/hr.2012.231
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hr.2012.231
Keywords
This article is cited by
-
Incidence and characteristics of spontaneous platelet macro-aggregation in acute ischemic stroke
Journal of Thrombosis and Thrombolysis (2021)
-
Mitochondrial coupling factor 6 upregulation in hypertension-induced cardiac hypertrophy
Herz (2015)
