
Abstract
A large number of congenital disorders are very rare and localized to rural areas in India, a country that practices both endogamy and consanguinity. Recent advances in genomics can aid in the identification of causative genomic elements when exploring therapeutic interventions and developing neonatal screening to assign novel functions. Here, we report a novel loss-of-function mutation (p.Trp370*) in the HACE1 gene that is associated with a rare congenital neurodevelopmental disorder in a boy from a remote village in southern India.
Similar content being viewed by others

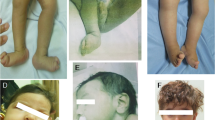
The proband came to Sri Sathya Sai Institute of Higher Medical Sciences, Prasanthigram, Puttaparthi (Andhra Pradesh, India) with a displaced fracture in the femoral neck following a fall and was found to have global developmental delay, severe mental retardation and hypotonia. The parents also reported multiple episodes of myoclonic seizures in the right upper and lower limbs at varying frequencies starting at 1 month after birth until ∼7 years of age. No generalized seizures were reported.
The child has had a significant delay in developmental milestones. The proband was able to sit and walk with support only at 6 years of age and had monosyllabic speech. The child is currently 11 years old, has better cognition, walks comfortably with support and has a broad-based, crouched gait. The patient has no ataxia or fasciculations. He is also able to get up from the ground with support. At present, the patient has hypertonia and exaggerated deep tendon reflexes in all four limbs with an extensor planter response in his feet bilaterally. The muscle power in the upper and lower limbs is normal. There is no wasting of muscles. The body hairs are hypopigmented, and the patient has heterochromia of the iris. Though the spine is clinically normal, exaggerated lumbar lordosis is present.
The child currently weighs 35 kg and is 130 cm tall. His weight is greater than the 25th percentile and his height less than the 3rd percentile of the age group as per the Indian Academy of Pediatrics growth chart. The occipitofrontal circumference is 53.5 cm and is within the range of normal. On the Vineland Social Maturity Scale, the child has a social age of 1 year and 7 months and a social quotient of 17. His current adaptive level falls below average, and the patient has a profound intellectual disability. His genitals appear normal, and his vision, hearing and dentition are normal. An electroencephalogram revealed an awake record background with well-defined α-activity at 9–10 Hz. The electroencephalogram also showed frequent bilateral temporal high voltage focal sharp waves and occasional generalized bilateral frontal spikes with wave discharges from the right frontal region.
The magnetic resonance imaging of the patient’s brain (Figure 1b) showed a hypoplastic corpus callosum, subtle soft tissue intensity lesion indenting the frontal horns of the lateral ventricle without blooming and lesion in the cerebrospinal fluid (that was partially inverted in the left premedullary cistern and displaced the basilar artery to the right with the presence of a possible neuroenteric cyst.
The boy with the neurodevelopmental disorder was born to consanguineous, healthy parents and has one unaffected older female sibling. There had been two male neonatal deaths (cause unknown) prior to the birth of the proband. An evaluation of the family tree revealed repeated consanguinity over two generations (Figure 1a). The family history of the patient gathered by the doctors from the parents of the proband revealed that the proband's uncle (III-7) was also affected with a similar disorder. The uncle himself never came to the hospital for any evaluation and did not give informed consent for genetic testing. Together, these findings strongly suggest the potential role of genetic inheritance in the disorder observed in the proband.

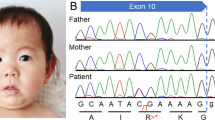
Radiological and molecular analysis of HACE1 (a) A family pedigree showing the proband (IV-5). (b) Magnetic resonance imaging (MRI; axial and sagittal views) of the proband showing cerebral atrophy, mega cisterna magna and a hypoplastic corpus callosum. (c) A comparison of the HACE1 (HECT domain and ankyrin repeat-containing E3 ubiquitin-protein ligase 1) sequence (from Sanger sequencing) of the proband to the reference (hg19:6:105177567–105177550). The 6:105233159.G>A mutation results in a loss of the HACE1 protein. (d) Western blot analysis to detect protein expression of HACE1 (sc-515746 antibody) reveals an absence of the 90 kDa protein in the proband (lane 1) and the presence of the same protein in the father (lane 2) and mother (lane 3). Appropriate protein loading controls are shown in the panel below. (e) The domain structure of the HACE1 protein with ankyrin repeats (green) and the HECT domain (red).17 The disorder-associated variants that have been previously reported are shown in purple, and the variant reported in this study is shown in pink.
Blood samples of the proband, his unaffected parents and siblings were collected after written informed consent from the parents. Standard karyotyping was conducted, and no abnormalities were found. The genomic DNA was then extracted from all five samples and were sequenced at the Institute of Bioinformatics and Applied Biotechnology (Bangalore, India). Whole-genome sequencing was performed on an Illumina HiSeq2500 platform (Illumina, San Diego, CA, USA) that generated 230 Gb of data with 100 bp paired-end reads. The average genome coverage was 17X-28X. The sequenced reads were aligned to the hg19 reference genome using bowtie2,1 and the alignment files were further processed with SAMtools.2 PCR duplicates were removed using Picard tools.3 The data were then pre-processed with IndelRealigner and Base Recalibrator tools from GATK.4 The variants from each sample were called separately using the HaplotypeCaller, followed by joint genotyping, variant quality score recalibration and genotype refinement according to GATK Best Practices workflow.5,6 The inherited homozygous variants from the proband were filtered out using the filter function from SnpSift7 and were annotated using SnpEff8 and SnpSift. Only the non-synonymous variants that had minor allele frequency <0.005 in the 1000G data set were considered for further analysis. The loss-of-function p.Trp370* mutation in HACE1 (HECT domain and ankyrin repeat-containing E3 ubiquitin-protein ligase 1) was shortlisted as a potentially causative mutation. The parents of the proband and the half-sister (Figure 1a) were heterozygous for this variant. In addition, the p.Trp370* variant in the HACE1 gene is not present in common variation databases, such as 1000G, dbSNP138, ExAC, UK10K, clinVar and HapMap, suggesting that this variation is unique to the family under study. This result was verified by Sanger sequencing and western blot analysis (Figure 1c,d).
HACE1 is involved in the subcellular localization and proteasomal degradation of target proteins and is known to be expressed in all regions of the brain and in many other tissues.9 HACE1 has been shown to regulate the activity of cellular GTPases, such as Rac1,10 which is believed to be involved in brain development.11 HACE1 is also reported to be a potential tumor suppressor. More specifically, it has been shown to be significantly downregulated in many human tumors.12,13
The case reported here is the 15th case worldwide and the first in India to report the involvement of the HACE1 gene in a neurodevelopmental disorder. In the first report, the study included three affected children from a German family with unrelated parents and five affected members from a Pakistani family with consanguinity. The five affected children from the Pakistani family had a homozygous deleterious mutation in the HACE1 gene, and the 3 children from the German family had compound heterozygosity in the same gene.14 In another large-scale study involving families affected with a neurodevelopmental disorder, the HACE1 mutation was reported in six affected children from four families, with one child each from three families with different homozygous loss-of-function mutations and three children from a single family with a biallelic mutation.15 Figure 1e and Table 1 list all of the HACE1 mutations reported thus far, including the p.Trp370* mutation described in the report.
In the OMIM (Online Mendelian Inheritance in Man) database, the disease assigned for the neurodevelopmental disorder caused by the HACE1 loss-of-function mutation is SPPRS (spastic paraplegia and psychomotor retardation with or without seizures, OMIM: 616756).16 The previously reported 14 cases of neurodevelopmental disorders with diverse loss-of-function mutations in the HACE1 gene are classified as SPPRS in the OMIM database. Given that neurodevelopmental disorders can display complex phenotypes, the case described here is likely another variation of SPPRS.
The diversity among the loss-of-function mutations in the HACE1 gene in a familial neurodevelopmental disorder from independent reports suggest that consanguinity and, perhaps, endogamy may play a significant role in propagating these deleterious autosomal recessive mutations through generations. This finding is of concern in cultures that practice endogamy and consanguinity as found in the case studied here. Developing an assay to screen for the mutation reported here to identify the prevalence of this mutation in the endogamous group that the family in this study belongs to is important. The assay can then be offered as a neonatal screen to this population.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
References
Langmead B, Salzberg SL . Fast gapped-read alignment with Bowtie 2. Nat Methods 2012; 9: 357–359.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009; 25: 2078–2079.
Broad Institute. Picard tools. Available at https://broadinstitute.github.io/picard/.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010; 20: 1297–1303.
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011; 43: 491–498.
Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics 2013; 43: 11.10.1–33.
Cingolani P, Patel VM, Coon M, Nguyen T, Land SJ, Ruden DM et al. Using Drosophila melanogaster as a model for genotoxic chemical mutational studies with a new program, SnpSift. Front Genet 2012; 3: 35.
Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012; 6: 80–92.
Anglesio MS, Evdokimova V, Melnyk N, Zhang L, Fernandez CV, Grundy PE et al. Differential expression of a novel ankyrin containing E3 ubiquitin-protein ligase, Hace1, in sporadic Wilms’ tumor versus normal kidney. Hum Mol Genet 2004; 13: 2061–2074.
Torrino S, Visvikis O, Doye A, Boyer L, Stefani C, Munro P et al. The E3 ubiquitin-ligase HACE1 catalyzes the ubiquitylation of active Rac1. Dev Cell 2011; 21: 959–965.
Mulherkar S, Uddin MD, Couvillon AD, Sillitoe RV, Tolias KF . The small GTPases RhoA and Rac1 regulate cerebellar development by controlling cell morphogenesis, migration and foliation. Dev Biol 2014; 394: 39–53.
Fernandez CV, Lestou VS, Wildish J, Lee CLY, Sorensen PHB . Detection of a novel t(6;15)(q21;q21) in a pediatric Wilms tumor. Cancer Genet Cytogenet 2001; 129: 165–167.
Zhang L, Anglesio MS, O’Sullivan M, Zhang F, Yang G, Sarao R et al. The E3 ligase HACE1 is a critical chromosome 6q21 tumor suppressor involved in multiple cancers. Nat Med 2007; 13: 1060–1069.
Hollstein R, Parry DA, Nalbach L, Logan CV, Strom TM, Hartill VL et al. HACE1 deficiency causes an autosomal recessive neurodevelopmental syndrome. J Med Genet 2015; 52: 797–803.
Akawi N, McRae J, Ansari M, Balasubramanian M, Blyth M, Brady AF et al. Discovery of four recessive developmental disorders using probabilistic genotype and phenotype matching among 4,125 families. Nat Genet 2015; 47: 1363–1369.
OMIM. Online Mendelian Inheritance in Man, OMIM®. McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University (Baltimore, MD). Available at http://omim.org/.
Jay JJ, Brouwer C . Lollipops in the clinic: information dense mutation plots for precision medicine. PLoS ONE 2016; 11: e0160519.
Data Citations
Srinivasan, Subhashini HGV Database http://dx.doi.org/10.6084/m9.figshare.hgv.1744 (2017)
Acknowledgements
The authors acknowledge Sri Sathya Sai Institute of Higher Medical Sciences, Prasanthigram, a super-specialty hospital in Puttaparthi, Andhra Pradesh, India, for providing completely free treatment to all patients and for introducing the family to one of the authors (BC). The authors also thank the GANIT lab for sequencing, IBAB for funding the sequencing and analysis efforts and DST-FIST SR/FST/LSI-536/2012 for an infrastructure grant. The Government of Karnataka provided funding for sequencing, and the Department of Science and Technology provided an infrastructure grant via DST-FIST (SR/FST/LSI-536/2012). The Department of Biosciences, SSSIHL, Prasanthi Nilayam, is being supported by DBT-BIF, UGC-SAP-DRS and DST-FIST, Government of India.
Author information
Authors and Affiliations
Contributions
NH performed the bioinformatic analyses and identified the disease and the causal variant. SR performed the wet lab validation. BC conceived the idea. BC and SS supervised the work. BEP, KNS and PK provided the samples and performed the clinical evaluation. NH, BC, SS and PK wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Hariharan, N., Ravi, S., Pradeep, B. et al. A novel loss-of-function mutation in HACE1 is linked to a genetic disorder in a patient from India. Hum Genome Var 5, 17061 (2018). https://doi.org/10.1038/hgv.2017.61
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2017.61
