
Abstract
G6PD deficiency is quite common in Italy where it is characterized by extreme molecular and biochemical heterogeneity. We report a 15-year-old Italian boy with G6PD Nilgiri (c.593G>A, p.Arg198His), a typical Indian variant of the Nilgiris tribal groups. Further, this variant was biochemically characterized, and the molecular screening of the family highlighted a de novo mutational event. To date, this family is the first Caucasian family carrying the G6PD Nilgiri variant.
Similar content being viewed by others


Although it is difficult to detect G6PD-deficient patients given that affected people are asymptomatic until they are exposed to triggers, >400 million individuals are thought to be G6PD-deficient, exhibiting high genetic heterogeneity and making this enzymopathy the most common clinically significant enzyme defect.1
G6PD deficiency occurs most frequently in Africa, Asia, the Mediterranean and the Middle East, synchronizing with endemic malaria.2 In fact, the prevalence of G6PD deficiency correlates with the geographical distribution of malaria, leading to the postulate that G6PD deficiency provides partial protection against this infection.3
To date, >200 different G6PD pathogenic variants have been identified worldwide, and each ethnic population exhibits a peculiar mutation profile. All G6PD variants are classified from Class I to V according to the residual enzyme activity and type of clinical manifestations.4,5
In 2008, Chalvam et al.6 reported the screening results for G6PD deficiency among 1,125 male individuals from different tribal groups of the Nilgiri district in Southern India. A hitherto unreported G6PD variant was identified in four individuals. This new variant (c.593G>A, based on NM_001042351.2, rs137852332) causes a predicted amino acid change of arginine (Arg) to histidine (His) at codon 198 in exon 6. This variant was confirmed by a family study and was designated G6PD Nilgiri.
Chalvam indicates that further studies must be performed to determine the prevalence and distribution of this variant in different population groups. Moreover, Chalvam et al. was unable to classify this variant given that a sufficient blood sample was not available for biochemical characterization of the residual enzyme. To date, G6PD Nilgiri has never been reported in the literature again.
In Italy, G6PD deficiency is characterized by extreme molecular and biochemical heterogeneity; in addition to Sardinia and Sicily, where higher disease prevalence is present (from 2 to 15%), G6PD-deficient subjects are also found in other Italian regions, such as Campania, Basilicata, Puglia and Lazio.7 All these regions presented endemic malaria in the past.
At least 10 distinct G6PD point variants have been reported in Italy,7 and >94% of those (based on NM_001042351.2 reference) include c.563C>T (G6PD Mediterranean), c.844G>C (G6PD Seattle) c.202G>A/c.376A>G (G6PD A−), c.1003G>A (G6PD Chatam) and c.1347G>C (G6PD Cassano). Thus, we are only able to perform preliminary mutational scanning of these variants.8 Moreover, novel and rare G6PD variants are also present.9
We report a case of an asymptomatic 15-year-old male born to parents of Italian descent (Campania region) with severe G6PD deficiency discovered during military recruiting (Table 1). He had no relevant family history. After written informed consent was provided, direct sequencing of the entire G6PD gene was performed given that the patient was negative for the most common Italian variants. The patient was hemizygous for the G6PD Nilgiri variant.
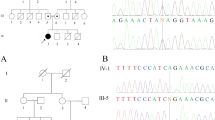
The family study confirmed the maternal inheritance of the variant (Figure 1a). Conversely, the p.Arg198His variant was not identified in the patient's grandparents, from whom DNA from buccal cell samples was analyzed (Figure 1b). The G6PD intragenic markers c.1365-13T>C (rs2071429) and c.1311C>T (rs2230037) were informative of the mother-grandparents relationships. These findings suggested that the G6PD Nilgiri variant in this family was a result of a de novo mutational event.

(a) The pedigree of the patient’s family is showed; (b) Sequences for the G6PD Nilgiri of proband and his relatives are showed. For the proband’s mother mutant and normal alleles were found in both hematopoietic and buccal cells. The absence of the c.593A allele in the grandparents of the proband shows a de novo occurrence of the G6PD Nilgiri in this family.
G6PD Nilgiri involves the same codon 198 that is mutated in G6PD Coimbra (c.592C>T, p.Arg198Cys). However, in G6PD Coimbra, the mutation is at nt 592, which is the first base of the codon, whereas the G6PD Nilgiri variant is located at the second base of the same codon. G6PD Coimbra is very close to the G6PD Mediterranean variant within exon 6 and has similar kinetic properties, namely high affinity for G6P and NADP+ and a high rate of deamino-nicotinamide adenine dinucleotide phosphate (dNADP) and 2-deoxy glucose-6-phosphate utilization compared with the G6PD-B enzyme (considered the normal phenotype). It has been suggested that the region encompassing the Coimbra and Mediterranean variants is spatially close and involved in the enzyme’s interactions with its substrate. Both variants are classified as Class II.4
This study reports for the first time the G6PD Nilgiri in Italy, and we were able to definitively classify this variant as Class II based on both on the residual enzyme activity and the clinical manifestations of the patient and his family members. In addition, this case provides further evidence on the prevalence and distribution of the G6PD Nilgiri in different population groups, confirming the high genetic heterogeneity of the G6PD deficiency in Italy,7 although owing to a de novo occurrence. In this context, we underscore c.592C>A (p.Arg198Ser)10 as an additional variant, indicating that the mutation at the same nucleotide site results from a de novo event; this consideration may suggest that this region is prone to such molecular events.
In conclusion, we highlight four main points: (a) a clinical picture of the patient and residual enzymatic activity are of primary importance for the definitive classification of each G6PD variant; (b) sequencing of the entire G6PD gene is mandatory especially in those patients from peculiar Italian regions; (c) the use of next-generation sequencing in G6PD-deficient subjects to diagnose common, rare and novel G6PD variants is now suggested,11 and finally, (d) G6PD is confirmed to be a gene prone to de novo events, as reported.12–17
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
References
Minucci A, Giardina B, Zuppi C, Capoluongo E . Glucose-6-phosphate dehydrogenase laboratory assay: how, when, and why? IUBMB Life 2009; 61: 27–34.
Allison AC . Malaria and glucose-6-phosphate dehydrogenase deficiency. Nature 1963; 197: 609–609.
Ruwende C, Hill A . Glucose-6-phosphate dehydrogenase deficiency and malaria. J Mol Med 1998; 76: 581–588.
Minucci A, Moradkhani K, Hwang MJ, Zuppi C, Giardina B, Capoluongo E . Glucose-6-phosphate dehydrogenase (G6PD) mutations database: review of the ‘old’ and update of the new mutations. Blood Cell Mol Dis 2012; 43: 154–165.
Gómez-Manzo S, Marcial-Quino J, Vanoye-Carlo A, Serrano-Posada H, Ortega-Cuellar D, González-Valdez A et al. Glucose-6-Phosphate Dehydrogenase: Update and Analysis of New Mutations around the World. Int J Mol Sci 2016; 9: 17.
Chalvam R, Kedar PS, Colah RB, Ghosh K, Mukherjee MB . A novel R198H mutation in the glucose-6-phosphate dehydrogenase gene in the tribal groups of the Nilgiris in Southern India. J Hum Genet 2008; 53: 181–184.
Martinez di Montemuros F, Dotti C, Tavazzi D, Fiorelli G, Cappellini MD . Molecular heterogeneity of glucose-6-phosphate dehydrogenase (G6PD) variants in Italy. Haematologica 1997; 82: 440–445.
Minucci A, Gentile L, Zuppi C, Giardina B, Capoluongo E . Rapid and simple identification of the commonest glucose-6-phosphate dehydrogenase (G6PD) Italian mutations: from DNA extraction to genotyping. Clin Chim Acta 2012; 14: 1018–1019.
Minucci A, Antenucci M, Giardina B, Zuppi C, Capoluongo E . G6PD Murcia, G6PD Ube and G6PD Orissa: report of three G6PD mutations unusual for Italian population. Clin Biochem 2010; 43: 1180–1181.
Warny M, Lausen B, Birgens H, Knabe N, Petersen J . Severe G6PD deficiency due to a new missense mutation in an infant of northern european descent. J Pediatr Hematol Oncol 2015; 37: e497–e499.
Bogari NM . Next generation sequencing (NGS) in glucose-6-phosphate dehydrogenase (G6PD) deficiency studies. Bioinformation 2016; 10: 41–43.
Zimmerman SA, Ware RE, Forman L, Westwood B, Beutler E . Glucose-6-phosphate dehydrogenase Durham: a de novo mutation associated with chronic hemolytic anemia. J Pediatr 1997; 131: 284–287.
Costa E, Cabeda JM, Vieira E, Pinto R, Pereira SA, Ferraz L et al. Glucose-6-phosphate dehydrogenase aveiro: a de novo mutation associated with chronic nonspherocytic hemolytic anemia. Blood 2000; 95: 1499–1501.
Van Wijk R, Huizinga EG, Prins I, Kors A, Rijksen G, Bierings M et al. Distinct phenotypic expression of two de novo missense mutations affecting the dimer interface of glucose-6-phosphate dehydrogenase. Blood Cells Mol Dis 2004; 32: 112–117.
Minucci A, Concolino P, Vendittelli F, Giardina B, Zuppi C, Capoluongo E . Glucose-6-phosphate dehydrogenase Buenos Aires: a novel de novo missense mutationassociated with severe enzyme deficiency. Clin Biochem 2008; 41: 742–745.
Jang MA, Kim JY, Lee KO, Kim SH, Koo HH, Kim HJ . A novel de novo mutation in the G6PD gene in a Korean boy with glucose-6-phosphate dehydrogenase deficiency: case report. Ann Clin Lab Sci 2015; 45: 446–448.
Del Orbe Barreto R, Arrizabalaga B, de la Hoz AB, Aragües P, Garcia-Ruiz JC, Arrieta A et al. Severe neonatal jaundice due to a de novo glucose-6-phosphate dehydrogenase deficient mutation. Int J Lab Hematol 2016; 38: e27–e29.
Data Citations
Minucci, Angelo HGV Database http://dx.doi.org/10.6084/m9.figshare.hgv.1738 (2017)
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Canu, G., Mazzuccato, G., Urbani, A. et al. Report of an Italian family carrying a typical Indian variant of the Nilgiris tribal groups resulting from a de novo occurrence. Hum Genome Var 5, 17057 (2018). https://doi.org/10.1038/hgv.2017.57
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2017.57
