
Abstract
Osteopetrosis is a heritable disorder of the skeleton that is characterized by increased bone density on radiographs caused by defects in osteoclast formation and function. Mutations in >10 genes are identified as causative for this clinically and genetically heterogeneous disease in humans. We report two novel missense variations in a compound heterozygous state in the CLCN7 gene, detected through targeted exome sequencing, in a 15-year-old Japanese female with intermediate autosomal recessive osteopetrosis.
Similar content being viewed by others


Osteopetrosis is a heritable, clinically, and genetically heterogeneous group of conditions that share the hallmark, observable on radiographs, of increased bone density caused by a failure of osteoclast development or function.1 The Nosology Group of the International Skeletal Dysplasia Society classifies these osteopetrotic conditions into several distinct entities based on clinical features, mode of inheritance, and underlying molecular and pathogenetic mechanisms.2 Mutations in at least 10 genes have been identified as causative in humans, accounting for 70% of all cases.3 These conditions can be inherited as autosomal recessive, dominant or X-linked traits, with the most severe forms being autosomal recessive. In addition to the paradigmatic recessive malignant and dominant benign forms, an intermediate group exists in which the defect can be inherited as either a recessive or a dominant trait. These intermediate cases often represent a diagnostic challenge, although diagnosis is mostly based on clinical and radiographic evaluation. Genetic testing can be used to confirm the diagnosis and to differentiate among the different subtypes of osteopetrosis, to provide additional information regarding the natural history and to predict likely responses to specific treatments and recurrence risks for adapted genetic counseling.1
Here, we present a case of intermediate autosomal recessive osteopetrosis (IARO; MIM 259710) with novel compound heterozygous mutations of the CLCN7 gene (MIM 602727), detected using a targeted exome sequencing (TES)-based approach to differentially cover possible causative genes for osteopetrosis, although IARO is generally caused by either homozygous/compound heterozygous mutations of CLCN7 or PLEKHM1.1
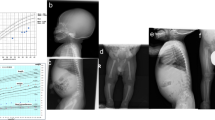
A 15-year-old Japanese female was the first child of non-consanguineous, healthy parents who had no notable family disease history (Figure 1a). Her first visit to the Department of Oral and Maxillofacial Surgery of Kobe University Hospital was at the age of 9 years. She had a history of fracture of the leg at the age of 5 years and was tentatively diagnosed with IARO based on clinical histories and signs at another hospital. Her early motor milestones and language development were normal, and she possessed normal intelligence. The patient presented with spontaneous fractures in the first years of life and a generalized increase in bone density. On presentation, her height and weight were 146.7 cm (−2.1 s.d.) and 50 kg (−0.2 s.d.), respectively. Her head circumference (55.9 cm) did not suggest any abnormality. She had normal hearing, but she had mild bilateral visual impairment due to atrophy of the bilateral optic nerves. Asymmetrical deformity of the fractured leg had not significantly progressed. She had a heavy malocclusion with hypodontia, several impacted teeth, enamel dysplasia, and malformed teeth (Figure 1b). Skeletal radiographs showed a diffuse increase in bone density with various characteristic signs, such as a ‘sandwich’ appearance in the vertebrae and a ‘bone-within-bone’ appearance of the iliac wings (Figure 1c–f). No hepatosplenomegaly was noted during the physical examination. Blood counts and phosphocalcic parameters were normal, and her karyotype was normal. No causative mutations were reported by sequence analyzes of all coding exons of TCIRG1 and CLCN7 performed at another hospital. Her clinical and radiological features were compatible with those of IARO, but the patient remained molecularly undiagnosed.

(a) Family pedigree; arrow shows the proband (P). (b) Orthopantomography showing impacted teeth, enamel hypoplasia and malformed premolars and roots. (c) Lateral radiograph of the skull showing diffuse thickening of the calvarium. (d) Lateral radiograph of the spine showing a ‘sandwich’ vertebrae appearance (arrows). (e) Radiograph of the lower limb and pelvis showing diffuse thickening of bones, with a ‘bone-within-bone’ appearance (arrows). (f) Radiograph of hands showing the ‘bone-within-bone’ appearance in metacarpals (arrows) and phalanges.
After informed consent was obtained from the patient’s parents, molecular diagnosis was performed on genomic DNA extracted from a blood sample. The study was approved by the Ethics Committees of Kobe University and Tokushima University. To simultaneously screen multiple osteopetrosis-causing genes, we first performed TES for the proband using a TruSight One Sequencing Panel (Illumina, San Diego, CA, USA), which allows the targeted sequencing of the exon regions of 4813 clinically relevant genes, and a MiSeq sequencer (Illumina) with our pipeline for next generation sequencing data analysis, as described elsewhere,4,5 with a minor modification due to a software update specific for a bioinformatics pipeline.6 To identify presumably pathogenic single-nucleotide variants, we excluded minor sequence variants with relatively high-allele frequencies, i.e., >0.01, included in the 1000 Genomes Project database (http://www.1000genomes.org/), the NHLBI GO Exome Sequencing Project (ESP6500, http://evs.gs.washington.edu/EVS/), the Human Genetic Variation Database (HGVD, http://www.genome.med.kyoto-u.ac.jp/SnpDB/) and the Integrative Japanese Genome Variation Database (iJGVD, https://ijgvd.megabank.tohoku.ac.jp/). The detection of copy-number variations (CNV) using TES data with a resolution of a single exon to several exons, depending on the exon size, was performed as previously described.6 These analyzes detected two different single-base substitutions, NM_001287.5(CLCN7_v001):c.1505G>A and c.1729G>A, in exons 17 and 19, respectively, and the substitutions in the exons were confirmed by Sanger sequencing (Figure 2a). The patient’s mother and father were heterozygous carriers of the c.1505G>A and the c.1729G>A variations, respectively, suggesting that these variations were present in the patient in a compound heterozygous state. These variations transformed the codon for cysteine 502 into that for tyrosine [NM_001287.5(CLCN7_i001):p.(Cys502Tyr)] and the codon for valine 577 into that for methionine [p.(Val577Met)], respectively. These variants are not present in the Human Gene Mutation Database (HGMD, Professional 2017.1, http://www.hgmd.org/) or ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/). No cases with p.(Cys502Tyr) are present in the publicly available variation databases, whereas one heterozygous case (carrier) with p.(Val577Met) was found in the Exome Aggregation Consortium (ExAC, http://exac.broadinstitute.org/) database. In multiple bioinformatics tools, including MutationTaster (http://www.mutationtaster.org/index.html), PolyPhen-2 version 2.2.2r405 (http://genetics.bwh.harvard.edu/pph2/index.shtml) and PROVEAN (http://provean.jcvi.org), p.(Cys502Tyr) was predicted to be deleterious by all three tools, whereas p.(Val577Met) was predicted to be deleterious by MutationTaster and Polyphen-2 and to be neutral by PROVEAN. p.(Cys502Tyr) and p.(Val577Met) are located within one of the helical transmembrane domains (TMDs) and within the loop between two TMDs, respectively. Multiple sequence alignment of CLCN7 orthologous proteins from different metazoan species revealed that these missense variants affected highly conserved residues (Figure 2b). No possible disease-causing variants, including CNVs, were detected within the potential candidate genes causing osteopetrosis in the TruSight One Sequencing Panel. As a result of the molecular diagnosis and re-evaluation of the clinical features of the affected patient and her unaffected parents having one of the mutations, the patient presented here was diagnosed with IARO caused by novel pathogenic missense mutations of CLCN7 in a compound heterozygous state. Because no possible features associated with osteopetrosis were observed in the patient’s sister, her allele status as a potential carrier was not determined through testing.

(a) Partial sequence chromatograms of exons 17 and 19 of CLCN7 in a patient and both parents. The nucleotide and corresponding amino acid sequences of the wild-type and mutant CLCN7 genes are also shown. (b) Multiple amino acid sequence alignment of the CLCN7 orthologous proteins from different metazoan species around the mutated amino acid. Arrowheads denote the mutated amino acid. Amino acids matching those in Homo sapiens are shaded.
Mutations in the CLCN7 gene encoding the chloride channel of the osteoclast membrane are associated with 75% of autosomal dominant osteopetrosis (ADO) cases, 10–15% of autosomal recessive osteopetrosis (ARO) cases and all cases of the intermediate form.7 Mutations in the CLCN7 gene affect the function of osteoclast-mediated extracellular acidification, resulting in the disturbed dissolution of the bone inorganic matrix and a series of clinical features.8 One of the mutations, p.(Val577Met), observed in the present case, was predicted to be a ‘neutral’ change by one of the three bioinformatics tools used in this study. With regard to the findings that the complete loss of function of the CLCN7 chloride channel causes malignant ARO, whereas dominant negative missense CLCN7 mutations are responsible for ADO,9 this result supports the hypothesis that the intermediate phenotype observed in IARO results from CLCN7 mutations that only mildly reduce the capacity of Cl− conductance.10 As noted by Pang et al.,11 a genetic analysis with prediction software would be insufficient to fully predict the functions of novel mutations in IARO due to the relatively small number of IARO cases and the possible partial residual capacity of Cl− conductance associated with mutated CLCN7 proteins in IARO. Our case contributes to enriching the database of CLCN7 mutations and improves our understanding of the clinical and biological aspects of IARO.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
References
Stark Z, Savarirayan R . Osteopetrosis. Orphanet J Rare Dis 2009; 4: 5.
Superti-Furga A, Unger S . Nosology and classification of genetic skeletal disorders: 2006 revision. Am J Med Genet A 2007; 143A: 1–18.
Sobacchi C, Schulz A, Coxon FP, Villa A, Helfrich MH . Osteopetrosis: genetics, treatment and new insights into osteoclast function. Nat Rev Endocrinol 2013; 9: 522–536.
Okamoto N, Naruto T, Kohmoto T, Komori T, Imoto I . A novel PTCH1 mutation in a patient with Gorlin syndrome. Hum Genome Var 2014; 1: 14022.
Watanabe M, Hayabuchi Y, Ono A, Naruto T, Horikawa H, Kohmoto T et al. Detection of 1p36 deletion by clinical exome-first diagnostic approach. Hum Genome Var 2016; 3: 16006.
Watanabe M, Nakagawa R, Naruto T, Kohmoto T, Suga K, Goji A et al. A novel missense mutation of COL5A2 in a patient with Ehlers-Danlos syndrome. Hum Genome Var 2016; 3: 16030.
Balemans W, van Wesenbeeck L, van Hul W . A clinical and molecular overview of the human osteopetroses. Calcif Tissue Int 2005; 77: 263–274.
Kornak U, Kasper D, Bosl MR, Kaiser E, Schweizer M, Schulz A et al. Loss of the ClC-7 chloride channel leads to osteopetrosis in mice and man. Cell 2001; 104: 205–215.
Cleiren E, Benichou O, Van Hul E, Gram J, Bollerslev J, Singer FR et al. Albers-Schonberg disease (autosomal dominant osteopetrosis, type II) results from mutations in the CLCN7 chloride channel gene. Hum Mol Genet 2001; 10: 2861–2867.
Campos-Xavier AB, Saraiva JM, Ribeiro LM, Munnich A, Cormier-Daire V . Chloride channel 7 (CLCN7) gene mutations in intermediate autosomal recessive osteopetrosis. Hum Genet 2003; 112: 186–189.
Pang Q, Chi Y, Zhao Z, Xing X, Li M, Wang O et al. Novel mutations of CLCN7 cause autosomal dominant osteopetrosis type II (ADO-II) and intermediate autosomal recessive osteopetrosis (IARO) in Chinese patients. Osteoporos Int 2016; 27: 1047–1055.
Data Citations
Imoto, Issei HGV Database http://dx.doi.org/10.6084/m9.figshare.hgv.1399 (2017)
Imoto, Issei HGV Database http://dx.doi.org/10.6084/m9.figshare.hgv.1402 (2017)
Acknowledgements
We thank the patient and her family for their participation in this study. We also thank our Orthopedics colleagues of Kobe University Hospital for their technical assistance. This work was partly performed in the Cooperative Research Project Program of the Medical Institute of Bioregulation, Kyushu University. This work was supported by the Japan Society for the Promotion of Science (16K15618, 15K19620, 26861783) and the Japan Agency for Medical Research and Development (16kk0205012h001, 16ek0109151h002).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Okamoto, N., Kohmoto, T., Naruto, T. et al. Novel CLCN7 compound heterozygous mutations in intermediate autosomal recessive osteopetrosis. Hum Genome Var 4, 17036 (2017). https://doi.org/10.1038/hgv.2017.36
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2017.36
This article is cited by
-
Genetics of Osteopetrosis
Current Osteoporosis Reports (2018)
-
Whole exome sequencing identified two novel homozygous missense variants in the same codon of CLCN7 underlying autosomal recessive infantile malignant osteopetrosis in a Pakistani family
Molecular Biology Reports (2018)
