
Abstract
Chromosomes of the siamang Symphalangus syndactylus (a small ape) carry large-scale heterochromatic structures at their ends. These structures look similar, by chromosome C-banding, to chromosome-end heterochromatin found in chimpanzee, bonobo and gorilla (African great apes), of which a major component is tandem repeats of 32-bp-long, AT-rich units. In the present study, we identified repetitive sequences that are a major component of the siamang heterochromatin. Their repeat units are 171 bp in length, and exhibit sequence similarity to alpha satellite DNA, a major component of the centromeres in primates. Thus, the large-scale heterochromatic structures have different origins between the great apes and the small ape. The presence of alpha satellite DNA in the telomere region has previously been reported in the white-cheeked gibbon Nomascus leucogenys, another small ape species. There is, however, a difference in the size of the telomere-region alpha satellite DNA, which is far larger in the siamang. It is not known whether the sequences of these two species (of different genera) have a common origin because the phylogenetic relationship of genera within the small ape family is still not clear. Possible evolutionary scenarios are discussed.
Similar content being viewed by others


Introduction
The telomere is a DNA-protein complex present at chromosome ends, and its DNA portion is known to comprise tandem repeats (called the telomere repeats) of a short unit, which is TTAGGG in the case of vertebrates (Meyne et al., 1989). Chromosomal regions immediately adjacent to telomeres often contain other repetitive sequences, which vary in their degree of repetitiveness (Brown et al., 1990; Cross et al., 1990; Weber et al., 1990). Because of the repetitive nature of these components, the telomere regions of humans are considered likely to form constitutive heterochromatin. However, their sizes are not large enough to be detected by chromosome C-banding on metaphase spreads. In contrast, chromosomes of African great apes carry easily detectable large heterochromatic structures at their ends (Yunis and Prakash, 1982). The main components of these structures are repetitive sequences called subterminal satellite (StSat) repeats that consist of 32-bp-long, AT-rich repeat units (Royle et al., 1994). Fluorescent in-situ hybridization (FISH) analysis of an StSat repeat probe to metaphase chromosomes revealed that the repeats are present in the majority of chimpanzee and bonobo chromosomes and in all chromosomes of the gorilla (H Hirai, unpublished results). We have previously estimated the total size of the StSat repeats to be as large as 0.1% of the chimpanzee genome (Koga et al., 2011). The distribution of the StSat repeats among the hominid species (human and African great apes) is somewhat puzzling because humans are phylogenetically closer to chimpanzees and bonobos than to gorillas. A question that readily arose from this patchy distribution was whether the StSat repeats were generated independently in the gorilla lineage and the chimpanzee/bonobo lineage, or whether the repeats were already present in the common ancestor of these great apes and were subsequently lost in the human lineage. We have recently settled this question (Koga et al., 2011): the latter explanation is more likely to be correct because the pattern of within-species variation is similar between gorilla and chimpanzee/bonobo, and this similarity is thought to stem from variation already present in the common ancestor. The molecular and cellular mechanisms of the emergence and disappearance of these structures have yet to be elucidated. Similar examples, if any, in the same or related taxa are expected to contribute to elucidating these mechanisms.
There is another example in the Hominoidea superfamily: we have previously found that chromosomes of the siamang (Symphalangus syndactylus (SSY)) carry large-scale constitutive heterochromatin at their ends (Wijayanto et al., 2005). This species is a small ape inhabiting the Malay peninsula and Sumatera island in South East Asia. ‘Small apes’ is a general name for the family Hylobatidae. In the currently most widely accepted primate taxonomy, this family and the families Pongidae and Hominidae form the superfamily Hominoidea. The African great apes belong to the family Hominidae. The C-band staining pattern of the chromosome-end heterochromatin looks similar between siamang and chimpanzee, but FISH analysis of an StSat probe to siamang chromosomes did not yield a positive signal (shown below). A genome sequence database is not available for this species. Therefore, relying upon the large size of these heterochromatin structures (and hence possibly a large amount of DNA), we cloned their constituent DNA by a method modified from the array comparative genomic hybridization technique. The species used for reference was the agile gibbon Hylobates agilis (HAG), another small ape that does not have large-scale heterochromatin in the telomere region. The results of sequencing analysis of the clones obtained from siamang were surprising: the telomere region contains the alpha satellite DNA that is a repetitive sequence known to be the primary DNA component of the centromere. Thus, the large-scale heterochromatic structures have different origins between the great apes and the small ape.
Materials and methods
Animals for collection of cells and DNA
The animals we used in the present study were chimpanzee Pan troglodytes (an adult male named Culleo who was born in Kyoto University Primate Research Institute), siamang SSY (a female infant stillborn in Hirakawa Zoo), agile gibbon HAG (an adult male bred at Kyoto University Primate Research Institute), a hybrid individual (a female born in Ishikawa Zoo) of white-handed gibbon Hylobates lar (HLA; female parent) and white-cheeked gibbon Nomascus leucogenys (NLE; male parent), and human Homo sapiens (an adult male donor). For the species of the Hylobatidae family, we hereafter use the three-letter abbreviations shown in the parentheses.
Chromosome preparation and FISH analysis
We cultured white blood cells, made chromosome preparations, and conducted C-band staining as previously described (Hirai et al., 1999, 2002). We performed FISH analysis designed for signal detection using three probes with three different colors (red, green and yellow) in a single assay, basically following the procedures in our previous work (Hirai et al., 1999, 2002, 2005). The labeling and detection substances for red color were biotin (using BioNick Labeling System; Life Technologies Inc., Carlsbad, CA, USA) and avidin-rhodamine conjugate, respectively. Those for green were digoxigenin (using DIG-Nick Translation Mix; Roche (Basel, Switzerland)) and anti-digoxigenin-FITC conjugate, respectively. For yellow color, we labeled the cloned DNA with biotin and digoxigenin separately, mixed the labeled DNAs, and then used the mixture as a single probe. The chromosome samples after hybridization were compiled into FISH image data using an AxioPlan 2 microscope (Carl Zeiss Inc., Jena, Germany), a Cool SNAP HQ camera (Photometrics, Tucson, AZ, USA), and an IPLab Spectrum image analyzer (Scanalystic Inc., Rockville, MD, USA). The stringency of hybridization can be considered to be moderate because the hybridization conditions described above are similar to those widely used for FISH analyses of mammalian chromosomes.
Preparation of genomic library
We collected genomic DNA from cultured fibroblast cells of SSY and HAG by a standard method (lysis with SDS, digestion of proteins with proteinase K, salt sedimentation and then isopropanol precipitation). A genomic library was prepared using a pCC1FOS Fosmid Library Construction Kit (Epicentre Biotechnologies, Madison, WI, USA) with some non-essential modifications to the protocol supplied by the manufacturer, as described in Koga et al. (2007). The vector was the 8.1-kb fosmid pCC1FOS, and the insert DNA was 35- to 45-kb genomic DNA fragments that had been mechanically sheared and recovered from an agarose gel piece after electrophoresis.
Screening by array comparative genomic hybridization
The purpose of this step was to identify DNA fragments that are highly repetitive in the SSY genome but not in the HAG genome. We first cultured 192 single colonies from the SSY genomic library in liquid medium distributed into wells of two 96-well plates. For each plate, we dotted two nylon membranes (as duplicates) with 1–2 μl of the bacterial cultures. We then performed hybridization experiments with the two sets of membranes (two membranes each) at the same time, by using an AlkPhos Direct Labelling and Detection System (GE Healthcare, Little Chalfont, UK). Genomic DNA of SSY was mechanically sheared to an approximate median size of 20 kb, and used as probe for one set of the membranes. The other set was hybridized with sheared genomic DNA of HAG. Comparing the autoradiograms obtained from the two sets, we selected clones that exhibited intense signals against the SSY genomic DNA but not against the HAG genomic DNA.
Other molecular techniques
We carried out subcloning, DNA sequencing and PCR, using the same methods as those in our previous work (Koga et al., 2006, 2007). Specific conditions are described below in each case.
Results
Constitutive heterochromatin at chromosome ends
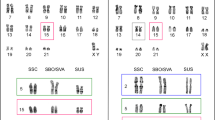
Figure 1 shows the results of C-band staining of metaphase spreads of human, chimpanzee, SSY, HAG and the HLA/NLE hybrid. Constitutive heterochromatin at chromosome ends was clearly observed with the samples of chimpanzee (the majority of the chromosomes) and SSY (all chromosomes), but not detectable with the other species examined.

Detection of heterochromatin by C-band staining. (a) Human; (b) chimpanzee; (c) SSY; (d) HAG; and (e) HLA/NLE hybrid. The bar in (a) represents 5 μm. Heavily stained regions contain heterochromatin.
Cloning of highly repetitive sequences
By array comparative genomic hybridization, we screened the SSY genomic library for fosmid clones containing DNA fragments that are highly repetitive in SSY but not in HAG. Figure 2 shows part of the autoradiograms obtained. There is one prominent spot in the left panel (with SSY genomic DNA as probe), and the signal intensity of the corresponding spot in the right panel (with HAG genomic DNA as probe) is about the same as that of other spots. This signal pattern indicates that this particular clone contains a DNA fragment highly repetitive only in SSY. Of the 192 clones screened, 4 exhibited this signal pattern.

An example of autoradiograms for isolation of species specific highly repetitive sequences by array comparative genomic hybridization. The nylon membrane corresponding to the left panel carried different clones picked up from an SSY genomic library, and was hybridized with SSY genomic DNA labeled with alkaline phosphatase as probe. The nylon membrane for the right panel contained the same set of the clones, prepared as a duplicate, and hybridized with genomic DNA of HAG. The upper-left part of the autoradiogram obtained from each membrane is shown here. The signal strength reflects the repetitiveness of each clone: the higher the copy number in the genome, the more intense the signal. The pattern of the signal strength among the clones is roughly the same between the two panels, except for one at the B-4 position. The clone at this position was assumed, and subsequently demonstrated, to be highly repetitive in the SSY genome but not in the HAG genome.
Nucleotide sequence of terminal regions
Because the pCC1FOS vector contains the lacZ gene and the cloning site is embedded there, the M13 universal primers (both the forward and reverse orientations) can be used for sequencing of terminal regions of the cloned fragments. We determined the sequences of 500–800 nucleotides from the ends of the 4 clones. All of the eight sequence reads were found to contain repetitive sequences consisting of repeat units of about 170 bp in length. All repeat units exhibited >75% nucleotide identity with one another. Using the respective sequences as queries, we conducted BLAST searches against all entries of the GenBank files. Every search resulted in a list of numerous hits to the alpha satellite DNA of primates (according to descriptions in the files).
Structure of a genomic clone
The genomic clones we obtained were all 35- to 45-kb-long fragments. We selected one clone at random (designated as pFosSia1) and used it for further analyses. Figure 3 shows the gel electrophoresis of this clone after digestion with restriction endonuclease BamHI (to completion) and/or Aor51HI (to varying extents). Because the fosmid vector pCC1FOS has two BamHI sites that bracket the cloning site, digestion of a fosmid clone with BamHI can separate the vector (8.1 kb) and insert portions. An analysis of the end-region sequences of pFosSia1 had suggested the occurrence of Aor51HI sites at intervals of about 170 bp.

Repetitive sequence structure revealed by restriction enzyme digestion. The second lane from the left (complete digestion with BamHI) shows two DNA fragments, the lower (8.1 kb) and upper (∼40 kb) bands being the vector (pCC1FOS) and insert (genomic DNA fragment of siamang), respectively. The third lane (complete digestion with BamHI and Aor51HI) contains two bands from the vector (split into two fragments of 4.2 and 3.9 kb due to an internal Aor51HI site) and other small fragments originating from the insert DNA. The prominent band at about the 0.2-kb position, which is absent in the second lane, indicates that the insert DNA digested with the two enzymes consists of a large number of restriction fragments (generated by Aor51HI digestion) of this size. The other two lanes contain the products of partial digestion with Aor51HI, and the appearance of ladder patterns indicates the presence of tandemly repeated sequences in the insert DNA.
Digestion of the fosmid clone with only BamHI produced a band the size of the vector and a single larger band, indicating that the insert portion (38 kb according to a subsequent pulsed-field electrophoresis analysis) does not have a BamHI site. Digestion with BamHI and a relatively large amount of Aor51HI yielded, in addition to two bands for the vector (split into 4.2-kb and 3.9-kb fragments due to an internal Aor51HI site), a bright band of about 170 bp and additional bands at locations of about 340, 510 and 680 bp. These results suggested that a sequence of about 170 bp in which one Aor51HI site resides is the basic repeat unit, that there are occasional nucleotide changes that eliminate the Aor51HI site, and that the repeat sequences cover the entire insert portion of the genomic clone. The long ladder patterns in the lanes for partial digestion with Aor51HI provide further evidence for the repetitive-sequence structure. The estimated size of the insert portion (38 kb) indicates that >200 repeat units are tandemly repeated in this clone. The repeat sequence that constitutes pFosSia1 was designated as SiaRep.
Consensus sequence and variation
Next, we determined the nucleotide sequence of the insert DNA of pFosSia1. Primer walking and shot-gun fragmentation are the principal strategies for obtaining the sequence of a long DNA fragment. These methods were, however, not adequate for pFosSia1 because of its repetitive complexity. We therefore employed a strategy that relies on partial digestion with Aor51HI.
We first transferred a terminal 8.0-kb region of the pFosSia1 insert (delimited by one BamHI site on the vector and an internal EcoRI site we found in a subsequent analysis) to plasmid pBluescript II SK+. We digested this subclone completely with EcoRV (one site in the vector but no site in the insert DNA) and then incompletely with Aor51HI. After ligation to form circular DNA molecules, we introduced the treated DNA into competent bacterial cells, spread the cells on plates and picked up several colonies. We selected plasmid clones whose lengths differed by multiples of about 510 bp (corresponding to three repeat units), sequenced them using the M13 universal primer, and edited them into a single stretch. The sequence finally obtained contained 24 consecutive repeat units.
We aligned the 24 repeat units manually, and determined a consensus sequence. The consensus sequence was defined as a collection of bases that occupied >50% of the corresponding nucleotide sites among the repeat units. The letter N was assigned to nucleotide sites in which the frequency of the most common base was 50% or less. The entire sequence of the consecutive 24 repeat units was deposited in GenBank (Accession number AB678729), and is shown in Figure 4. The distribution of the repeat lengths was 170 bp (6/24; 25%), 171 bp (16/24; 67%), 172 bp (1/24; 4%) and 173 bp (1/24; 4%). The average pairwise sequence identity, calculated by excluding insertions and deletions, was 86%.

The entire nucleotide sequence of the 24 repeat units in pFosSia1. ‘Con’ indicates the consensus sequence. Nucleotide sites occupied by the same base as that in the consensus sequence are indicated by dots. Nucleotide sites containing different bases are shown by the respective bases observed. The minus symbol implies absence of a nucleotide at its position. The asterisk on the consensus sequence indicates that all the 24 repeat units have the same nucleotide at the indicated position.
Comparison with sequences of alpha satellite DNA
We compared the consensus sequence of the repeat units of pFosSia1 with the consensus sequence of alpha satellite DNA of human (Alexandrov et al., 1993), orangutan (Haaf and Willard, 1998) and NLE (Cellamare et al., 2009), which is, to our knowledge, the only Hylobatidae species whose alpha satellite DNA has been extensively analyzed at the nucleotide sequence level. Figure 5 shows the alignment among these species we made manually. The SSY consensus sequence exhibits 92% identity with the consensus sequence of NLE, and about 80% identity with those of the two Hominidae species.

Comparison of the consensus sequence of the repeat unit in pFosSia1 and those of alpha satellite DNA of three other primate species. ‘Ora’ and ‘Hum’ are abbreviations for orangutan and human, respectively. The sequence identity to the SSY consensus sequence is shown after the sequence of each species.
FISH analysis for chromosomal locations
We conducted FISH analysis of mitotic metaphase chromosomes for chromosomal locations of SiaRep, the StSat repeats (the main component of the chimpanzee subtelomeric heterochromatin) and 18S ribosomal DNA. The last probe served as a positive control for the hybridization and detection processes. For each species (or a hybrid animal), we spread cells on a slide glass, used 10–30 complete metaphase spreads for examination of hybridization patterns, and verified that the patterns observed were virtually identical among these chromosome sets. As shown in Figure 6, 18S ribosomal DNA (yellow) showed nine signals in human (panel a), eight in chimpanzee (panel b), two (closely located on this chromosome spread) in SSY (panel c), two in HAG (panel d) and three in the HLA/NLE hybrid (panel e). These numbers are in accord with those expected for the respective species (Hirai et al., 1999).

FISH analysis of chromosomes for locations of the repeat sequences. Three probes were used in single hybridization assays: StSat repeats (green), SiaRep (red) and a clone of human 18S ribosomal DNA (yellow; see Hirai et al., 1999). The last probe served as a positive control for hybridization reactions. (a) Human; (b) chimpanzee; (c) SSY; (d) HAG and (e) HLA/NLE hybrid. The bar in (a) represents 10 μm. Panel (f) is not a photograph of fluorescence detection but a DAPI banding pattern of the chromosome spread used for panel (e). This treatment yields G band-like bands, and enables, based on the chromosome shape and banding patterns, identification of the origin (HLA or NLE) and the chromosome number (Hirai et al., 2007). Chromosomes originating from the HLA parent are marked with white dots. Chromosomes without dots are those derived from the NLE parent. Scanning of chromosome spreads for (a–e) using an image analyzer was first conducted at its default settings that automatically achieved the highest signal-to-noise ratio. Photographs in (a, b, d and e) were those obtained with these settings. We then scanned the same spreads again at a manual setting to attain higher sensitivity (and a larger amount of noise at the same time). The siamang sample exhibited signals only in the telomere regions in the first scan, but the second scan detected additional faint signals in centromere regions. Panel (c) is a photograph obtained in this second scan. No additional signals were found in the other four samples. In (c), some, but not all, relatively strong signals in the centromere regions are indicated by arrowheads. The black-and-white photograph overlaid in (c) was produced by a further scan of the right part of the chromosome spread for the luminance level due only to biotin-rhodamin (labeling substance for the SiaRep probe).
With the SiaRep probe (red), the heterochromatin in the telomere region of all SSY chromosomes yielded intense hybridization signals, indicating that SiaRep is a major DNA component of these heterochromatic structures (panel c). In addition to these signals in the telomere regions, faint signals were observed in centromere regions of about half of the chromosomes. In contrast, signals were observed only in centromere regions of all chromosomes in HAG (panel d). In the sample of the HLA/NLE hybrid (panel e), chromosomes originating from the NLE parent all exhibited signals in both the telomere regions and centromere regions, while signals were observed in only the centromere regions on the chromosomes from the HLA parent. The relative strength of the signals of the telomere regions to those of the centromere regions clearly differed between the SSY chromosomes and NLE chromosomes, being higher in the former. The samples of human and chimpanzee did not exhibit detectable signals.
For the StSat repeats (green), as expected, chimpanzee chromosomes yielded clear signals and human chromosomes did not. No positive signal was observed in the Hylobatidae samples.
Discussion
Origins of large-scale heterochromatin structures in different taxa
The StSat repeats (tandem repeats of 32-bp units) constitute the subtelomeric heterochromatin of chimpanzee (and other African great apes), but are not likely to be a component of that of SSY. SiaRep repeats (tandem repeats of 171-bp units) make up the SSY telomere-region heterochromatin, and appear not to exist in that of chimpanzee. It has not been confirmed whether these two repeat sequences are the most abundant DNA components of the respective heterochromatin structures. They are, however, present there at least as ubiquitous components of the respective structures because all the chromosome ends where large heterochromatin was observed by C-banding (Figure 1) exhibited positive signals in the FISH analysis (Figure 6). The consensus sequences of these two repeats do not share a similar nucleotide block recognizable by visual inspection. It can thus be inferred that the large-scale heterochromatic structures have different origins between the great apes and the small ape.
One feature common of the two types of repeats is the content of the A and T residues in their consensus sequences: the content (calculated without including polymorphic nucleotide sites) is 65% (20/31) in StSat and 65% (108/169) in SiaRep. The amount of AT content might be a significant factor for expansion and/or maintenance of these structures, but the fact that the same amount was observed may be just a coincidence.
Presence of alpha satellite DNA in the telomere region
Alpha satellite DNA is a major DNA component of primate centromeres (Willard, 1991; Alves et al., 1994; Lee et al., 2011). In the family Hominidae, in addition to occurring in the centromeric heterochromatin, alpha satellite DNA occurs in some interstitial regions, which are thought to be remnants of once-functional centromeres (Reddy and Sulcova, 1998). To our knowledge, however, alpha satellite DNA has not been found in telomere regions in Hominidae. In the family Hylobatidae, one such example has been reported: alpha satellite DNA in telomere regions of the chromosomes of NLE (Cellamare et al., 2009). In the present study, we confirmed this and detected another example (SSY) within Hylobatidae. Our results are, however, not simply a second example. The present results of chromosome C-banding and FISH analysis demonstrate that alpha satellite DNA has expanded to large-scale heterochromatin blocks in the lineage leading to the SSY, regardless of whether they have a common origin. Differences in the sizes of the StSat repeats and other telomere-region repetitive sequences have also been reported in great apes (between chimpanzee and gorilla) (Ventura et al., 2011). The mechanisms leading to these size differences are not known. Comparative studies between the great apes and small apes are expected to be a powerful approach to elucidate the mechanisms.
Besides the amplification mechanisms, the mechanism of first occurrence of the alpha satellite DNA in the telomere region is of great interest. One possibility would be that a transposable element, or virus, mediated its migration from the centromere to the telomere region. Some transposable elements are known to often carry their 5′- or 3′-flanking regions upon transposition (Goodier et al., 2000; Pickeral et al., 2000; Xing et al., 2006). Another possibility would be an event similar to that giving rise to the alpha satellite DNA found in interstitial regions of Hominidae (mentioned above). It is well known that species of the Hylobatidae family have undergone frequent chromosomal reorganizations, including fissions, fusions and translocations of chromosomes (Jauch et al., 1992; Mueller et al., 2003). A fission of a chromosome at the centromere, followed by telomere formation at the breakpoint, might have been the origin of alpha satellite DNA in the telomere region.
History of telomere-region alpha satellite DNA in the Hylobatidae family
In the latest Hylobatidae taxonomy, this family consists of four genera (Symphalangus, Hylobates, Nomascus and Hoolock). The phylogenetic relationship of these genera is still not clear probably because of the relatively short time span in which their lineages diverged, and some additional factors such as gene flow among species (Van Ngoc et al., 2010; Kim et al., 2011). In our discussion below, we consider three of them (Symphalangus, Hylobates and Nomascus) because the materials we used in the present study were from species of these three genera.
The intensity of FISH signals in the telomere regions was strong for SSY (Symphalangus), relatively weak for NLE (Nomascus) and no signal was observed in telomere regions for HAG or HLA (Hylobates). We designate these situations as ++, + and 0, respectively, and define the transition from one situation to another (generation, amplification, reduction and extinction) as shown in Figure 7. Here, we assume that change from 0 to ++ always passes through +. We assume, however, that a direct change from ++ to 0 can happen because such a change is likely to have occurred in the human lineage with the StSat repeats (Koga et al., 2011). One possible mechanism we have in mind is truncation of chromosomes and regeneration of the telomere.

Patterns of situation changes that include relatively small numbers (up to three) of events. Three kinds of divergence patterns (a, b, c, d; e–h) of the three genera (Nomascus, Symphalangus and Hylobates) are considered. The signs in parentheses show the relative strength of FISH signals in the telomere region (+ and ++) and absence of signals (0). Four kinds of transitions were defined as shown at the bottom, and designated with their first letters (underlined). The letters are placed on branches where they are assumed to have occurred in each branching pattern.
There are three possible topologies for branching patterns of three genera, depending on which genus is assumed to have diverged first. Possible scenarios that involve three or fewer transition events are the eight cases shown in Figure 7 (a and b, first divergence of Nomascus; c and d, Symphalangus first; e–h, Hylobates first). To narrow these down, it is necessary to wait for accumulation of more knowledge about the phylogeny of the Hylobatidae family. There is, however, a possibility that surveying more species for the presence/absence, and the size if present, of the telomere-region alpha satellite DNA would contribute to clarifying the phylogenetics of the Hylobatidae family. For example, the results so far obtained appear to give a little stronger support to the hypothesis of the most recent divergence of Nomascus and Symphalangus because case (e) requires the smallest number (two) of events.
Data Archiving
Consensus sequence data have been deposited in the GenBank database under accession number AB678729.
Accession codes
References
Alexandrov IA, Medvedev LI, Mashkova TD, Kisselev LL, Romanova LY, Yurov YB (1993). Definition of a new alpha satellite suprachromosomal family characterized by monomeric organization. Nucleic Acids Res 21: 2209–2215.
Alves G, Seuánez HN, Fanning T (1994). Alpha satellite DNA in neotropical primates (Platyrrhini). Chromosoma 103: 262–267.
Brown WR, MacKinnon PJ, Villasanté A, Spurr N, Buckle VJ, Dobson MJ (1990). Structure and polymorphism of human telomere-associated DNA. Cell 63: 119–132.
Cellamare A, Catacchio CR, Alkan C, Giannuzzi G, Antonacci F, Cardone MF et al (2009). New insights into centromere organization and evolution from the white-cheeked gibbon and marmoset. Mol Biol Evol 26: 1889–1900.
Cross S, Lindsey J, Fantes J, McKay S, McGill N, Cooke H (1990). The structure of a subterminal repeated sequence present on many human chromosomes. Nucleic Acids Res 18: 6649–6657.
Goodier JL, Ostertag EM, Kazazian HH (2000). Transduction of 3'-flanking sequences is common in L1 retrotransposition. Hum Mol Genet 9: 653–657.
Haaf T, Willard HF (1998). Orangutan alpha-satellite monomers are closely related to the human consensus sequence. Mamm Genome 9: 440–447.
Hirai H, Hirai Y, Domae H, Kirihara Y (2007). A most distant intergeneric hybrid offspring (Larcon) of lesser apes, Nomascus leucogenys and Hylobates lar. Hum Genet 122: 477–483.
Hirai H, Hirai Y, Kawamoto Y, Endo H, Kimura J, Rerkamnuaychoke W (2002). Cytogenetic differentiation of two sympatric tree shrew taxa found in the southern part of the Isthmus of Kra. Chromosome Res 10: 313–327.
Hirai H, Matsubayashi K, Kumazaki K, Kato A, Maeda N, Kim HS (2005). Chimpanzee chromosomes: retrotransposable compound repeat DNA organization (RCRO) and its influence on meiotic prophase and crossing-over. Cytogenet Genome Res 108: 248–254.
Hirai H, Taguchi T, Godwin AK (1999). Genomic differentiation of 18S ribosomal DNA and beta-satellite DNA in the hominoid and its evolutionary aspects. Chromosome Res 7: 531–540.
Jauch A, Wienberg J, Stanyon R, Arnold N, Tofanelli S, Ishida T et al (1992). Reconstruction of genomic rearrangements in great apes and gibbons by chromosome painting. Proc Natl Acad Sci USA 89: 8611–8615.
Kim SK, Carbone L, Becquet C, Mootnick AR, Li DJ, de Jong PJ et al (2011). Patterns of genetic variation within and between Gibbon species. Mol Biol Evol 28: 2211–2218.
Koga A, Iida A, Hori H, Shimada A, Shima A (2006). Vertebrate DNA transposon as a natural mutator: the medaka fish Tol2 element contributes to genetic variation without recognizable traces. Mol Biol Evol 23: 1414–1419.
Koga A, Notohara M, Hirai H (2011). Evolution of subterminal satellite (StSat) repeats in hominids. Genetica 139: 167–175.
Koga A, Shimada A, Kuroki T, Hori H, Kusumi J, Kyono-Hamaguchi Y et al (2007). The Tol1 transposable element of the medaka fish moves in human and mouse cells. J Hum Genet 52: 628–635.
Lee HR, Hayden KE, Willard HF (2011). Organization and molecular evolution of CENP-A-associated satellite DNA families in a basal primate genome. Genome Biol Evol 3: 1136–1149.
Meyne J, Ratliff RL, Moyzis RK (1989). Conservation of the human telomere sequence (TTAGGG)n among vertebrates. Proc Natl Acad Sci USA 86: 7049–7053.
Mueller S, Hollatz M, Wienberg J (2003). Chromosomal phylogeny and evolution of gibbons (Hylobatidae). Hum Genet 113: 493Y501.
Pickeral OK, Makalowski W, Boguski MS, Boeke JD : Frequent human genomic DNA transduction driven by LINE-1 retrotransposition. Genome Res 10: 411–415 (2000).
Reddy KS, Sulcova V (1998). The mobile nature of acrocentric elements illustrated by three unusual chromosome variants. Hum Genet 102: 653–662.
Royle NJ, Baird DM, Jeffreys AJ (1994). A subterminal satellite located adjacent to telomeres in chimpanzees is absent from the human genome. Nat Genet 6: 52–56.
Van Ngoc T, Mootnick AR, Li M, Ziegler T, Agil M, Moisson P et al (2010). Mitochondrial evidence for multiple radiations in the evolutionary history of small apes. BMC Evol Biol 10: 74.
Ventura M, Catacchio CR, Alkan C, Marques-Bonet T, Sajjadian S, Graves TA et al (2011). Gorilla genome structural variation reveals evolutionary parallelisms with chimpanzee. Genome Res 21: 1640–1649.
Weber B, Collins C, Robbins C, Magenis RE, Delaney AD, Gray JW et al (1990). Characterization and organization of DNA sequences adjacent to the human telomere associated repeat (TTAGGG)n. Nucleic Acids Res 18: 3353–3361.
Wijayanto H, Hirai Y, Kamanaka Y, Katho A, Sajuthi D, Hirai H (2005). Patterns of C-heterochromatin and telomeric DNA in two representative groups of small apes, the genera Hylobates and Symphalangus. Chromosome Res 13: 717–724.
Willard HF (1991). Evolution of alpha satellite. Curr Opin Genet Dev 1: 509–514.
Xing J et al. Emergence of primate genes by retrotransposon-mediated sequence transduction. Proc Natl Acad Sci USA 103: 17608–17613 (2006) This study shows the evolutionary significance of retrotransposon-mediated transduction during human evolution.
Yunis JJ, Prakash O (1982). The origin of man: a chromosomal pictorial legacy. Science 215: 1525–1530.
Acknowledgements
We are grateful to Drs Hiroshi Masumoto, Atsushi Matsui and Elizabeth Nakajima for helpful discussions, to Hirakawa Zoo for providing tissue samples of SSY through the Great Ape Information Network (GAIN) program and to Ishikawa Zoo for providing tissue samples of the HLA/NLE hybrid animal. This work was supported by Grants-in-Aid (23657165 to AK, 22247037 to HH and 20405016 to HH) and the Global COE program (A06 to Kyoto University) from the MEXT of Japan.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Koga, A., Hirai, Y., Hara, T. et al. Repetitive sequences originating from the centromere constitute large-scale heterochromatin in the telomere region in the siamang, a small ape. Heredity 109, 180–187 (2012). https://doi.org/10.1038/hdy.2012.28
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2012.28
Keywords
This article is cited by
-
Higher-order repeat structure in alpha satellite DNA occurs in New World monkeys and is not confined to hominoids
Scientific Reports (2015)
-
Higher-order repeat structure in alpha satellite DNA is an attribute of hominoids rather than hominids
Journal of Human Genetics (2013)
-
Tandem repeat sequences evolutionarily related to SVA-type retrotransposons are expanded in the centromere region of the western hoolock gibbon, a small ape
Journal of Human Genetics (2012)
