
Abstract
Purpose
The zone of polarizing activity regulatory sequence (ZRS) is an enhancer that regulates sonic hedgehog during embryonic limb development. Recently, mutations in a noncoding evolutionary conserved sequence 500 bp upstream of the ZRS, termed the pre-ZRS (pZRS), have been associated with polydactyly in dogs and humans. Here, we report the first case of triphalangeal thumb–polysyndactyly syndrome (TPT-PS) to be associated with mutations in this region and show via mouse enhancer assays how this mutation leads to ectopic expression throughout the developing limb bud.
Methods
We used linkage analysis, whole-exome sequencing, Sanger sequencing, fluorescence in situ hybridization, multiplex ligation-dependent probe amplification, single-nucleotide polymorphism array, and a mouse transgenic enhancer assay.
Results
Ten members of a TPT-PS family were included in this study. The mutation was linked to chromosome 7q36 (LOD score 3.0). No aberrations in the ZRS could be identified. A point mutation in the pZRS (chr7:156585476G>C; GRCh37/hg19) was detected in all affected family members. Functional characterization using a mouse transgenic enhancer essay showed extended ectopic expression dispersed throughout the entire limb bud (E11.5).
Conclusion
Our work describes the first mutation in the pZRS to be associated with TPT-PS and provides functional evidence that this mutation leads to ectopic expression of this enhancer within the developing limb.
Similar content being viewed by others


Introduction
The genomic landscape of chromosome 7q36 comprises a topological associated domain (TAD),1ranging from sonic hedgehog (SHH) to limb development membrane protein 1 (LMBR1), a gene located ~1 megabase (Mb) upstream of SHH.2 Within this TAD, several regulatory enhancer elements have been identified that are involved in SHH regulation during embryonic development of the brain, the central nervous system, and the limbs.3,4 In limb development, SHH expression is limited to the zone of polarizing activity (ZPA) in the posterior limb bud, creating an SHH gradient over the anteroposterior axis of the limb.5 This gradient is crucial for establishing adequate digit patterning.
The regulation of SHH in the embryonic limb bud has largely been attributed to a conserved noncoding regulatory element located in intron 5 of LMBR1, called the ZPA regulatory sequence (ZRS).6 Various genetic aberrations of the ZRS have been associated with triphalangeal thumb (TPT) and preaxial polydactyly in humans, mice, cats, and chickens.7,8 Molecular studies in polydactylous mice revealed that disruption of the ZRS results in ectopic SHH expression in the anterior limb bud.6
Point mutations and genomic duplications are the most commonly found ZRS aberrations that lead to limb malformations. Point mutations of the ZRS generally cause triphalangeal thumb accompanied with an additional thumb.9 Genomic duplications encompassing the ZRS are associated with more severe phenotypes, like triphalangeal thumb–polysyndactyly syndrome (TPT-PS), Haas-type polysyndactyly, or Laurin–Sandrow syndrome.10 To date, many point mutations of the ZRS and genomic duplications of different sizes have been reported in the literature.9,10
Recently, several studies have suggested that genetic alterations in locations other than the ZRS throughout the 1 Mb encompassing SHH-LMBR1 TAD could be associated with TPT phenotypes. Petit et al.1 showed that a 2-kilobase (kb) deletion in a gene desert 240 kb upstream of SHH was linked to familial TPT and hypertrichosis. Additionally, variations in the pre-ZRS (pZRS), a noncoding conserved region approximately 700 base pairs (bp) upstream to the ZRS, were reported in sporadic cases of preaxial polydactyly in humans and dogs.12,13
In 1988, Nicolai et al.14 reported a large Dutch TPT-PS family consisting of 27 patients. The family pedigree suggested an autosomal dominant inheritance. Subsequently, Tsukurov et al.15 linked the disease locus in this family to chromosome 7q36 (D7S550, logarithm of the odds (LOD) score 6.85). Here, we further investigated the molecular causes of this severe limb anomaly in this Dutch TPT-PS family. A whole-genome single-nucleotide polymorphism (SNP) array found linkage between the TPT-PS and the 7q36 locus and, combined with multiplex ligation-dependent probe amplification, did not identify any disease-associated copy-number variations (CNVs). Karyotyping and fluorescence in situ hybridization (FISH) did not reveal any specific structural rearrangements within this region. ZRS sequencing and whole-exome sequencing revealed no pathogenic variants. We next sequenced a 4,500-bp region around the ZRS and identified a point mutation in the pZRS that segregated with the phenotype. Computational analyses did not find increased conservation scores for this mutation compared with previously reported mutations that lead to a much less severe phenotype. A mouse transgenic enhancer essay showed ectopic LacZ expression dispersed through the entire limb bud at E11.5, in both fore- and hindlimbs. Combined, our study underlines that the integrity of the surrounding environment of the ZRS plays a crucial role in the regulation of SHH during limb development.
Materials and methods
TPT-PS family
Two TPT-PS families were identified at the outpatient clinic for Congenital Hand and Upper Limb Anomalies at the Sophia Children’s Hospital in Rotterdam, the Netherlands. The family members were consulted and clinically examined by a plastic surgeon and a clinical geneticist. A common ancestor for two Dutch TPT-PS families could be identified in the established pedigree, confirming relatedness of both families (Figure 1). Peripheral blood samples were collected from the affected father, unaffected mother, and both affected children in Dutch family I and the affected mother and child in Dutch family II. Additionally, the Dutch family reported the presence of Australian relatives with similar congenital hand anomalies. The Australian family was subsequently consulted and clinically examined and peripheral blood samples were collected from four affected family members in the Australian family. In total, nine patients with TPT-PS and one unaffected family member were included in this study. Written informed consent was obtained from all family members. This study has been approved by the Medical Ethics Committee of the Erasmus University Medical Centre in Rotterdam, the Netherlands (MEC-2015-12).

Pedigree of triphalangeal thumb–polysyndactyly syndrome (TPT-PS) family. Affected family members are identified by filled black squares or circles. The numbers of the 10 family members that were included in this study are underlined and in bold.
SNP array and linkage
Genome-wide genotyping was conducted using 200 ng DNA from all included family members with an Illumina Infinium GSAMD-24v1 array (Illumina, San Diego, CA; 730,525 SNPs at a median distance of 2.1 kb). The statistical package, easyLINKAGE Plus v5.08 (20), Merlin v1.0.1 software (Abecasis Lab, University of Michigan), was used to perform single-point and multipoint parametric linkage analysis as previously described.16 Logarithm of odds scores were obtained using a dominant model of inheritance, with 99% penetrance and disease allele frequency of 1:1.000. Furthermore, SNP-array data was additionally used to evaluate the presence of genomic duplications or deletions. Data was analyzed using the Nexus Copy Number, Discovery Edition, version 7 (BioDiscovery, El Segundo, CA).
ZRS sequencing and multiplex ligation-dependent probe amplification
Targeted sequencing of the ZRS combined with multiplex ligation-dependent probe amplification is currently used as standard genetic diagnostic work-up in TPT families. DNA was isolated from peripheral blood. Fragments were amplified using standard polymerase chain reaction. An 834-bp fragment covering the ZRS was sequenced in all Dutch index patients (chr7:156583766-156584600, GRCh/Hg19).
Additionally, primers were designed to sequence a 4,500-bp region surrounding the ZRS (chr7:15681430-156585993, GRCh39, Hg19). A total of eight primer pairs were required to cover this entire sequence (Supplementary Table S1 online).
The polymerase chain reaction products were sequenced using Big Dye Terminator 3.1. The fragments were loaded on an Abi 3130 sequence analyzer and genetic analysis was performed with SeqScape Software (v3.0).
Multiplex ligation-dependent probe amplification analysis (MRC Holland S134 kit) was used to detect the presence of genomic duplications and deletions between exons 3 and 6 of LMBR1, encompassing the ZRS.
Karyotyping and FISH
Karyotyping was performed on GTG-banded metaphases obtained from peripheral blood cultures using standard procedures. Karyotypes were obtained from V-6 and a control. Results were described in accordance with the International System for Human Cytogenetic Nomenclature 2005 (ref.17). FISH was carried out on the fixed sediments of the karyotyping cultures of subject V-6 and a control. FISH analysis was performed using five 7q36 located bacterial artificial chromosome (BAC) clones. The BAC clones (RP11-773c4, RP5-982e9, RP11-332e22, RP11-51l24, and RP11-50D7; Supplementary Table S2 online) were selected from the University of California–Santa Cruz Genome Browser (assembly March 2006) and ordered from BACPAC Resources (Children’s Hospital of Oakland Research Institute, Oakland, CA). After isolation of the BAC DNA, the probes were labeled and used for FISH on chromosome preparations from patients and parents, according to standard protocols.18
Whole-exome sequencing
Samples of patients IV-2, V-1, V-4, and V-5 were sequenced using an Illumina Nextseq500 machine. The exome samples were captured using the Truseq Exome Library Prep Kit and the Illumina TruSeq Exome Library Prep Reference Guide v01. Reads were aligned against the Human Reference Genome build 19 (hg19) and genetic variants were called using the Qiagen CLC genomics workbench. The variant call format (.vcf) files were annotated with Annovar.19 Subsequently, the annotated data was filtered using the previously obtained linkage data, the presence of the variant in all four members of the family, and an allele frequency of less than 0.001 in publicly available reference data sets, including 1000 Genomes,20 ExAC,21 and Qiagen HGMD Professional.22
Mouse enhancer assay
A genomic region encompassing both pZRS and ZRS (chr7:156583545-156585773, GRCh37/hg19, 2229 base pairs) with either the reference sequence or the chr7:156585476G>C change was cloned into an HSP 68-Lac Z vector (Addgene #37843) by carrying out a polymerase chain reaction on genomic DNA from patient V-5. The cloned plasmids were sequenced verified to contain the mutation allele and the wild-type allele and to exclude any other variants. Transgenic mouse E11.5 embryos followed by β-galactosidase staining were generated by Cyagen Biosciences. Pictures of embryos were taken using a Leica M205FA stereomicroscope and annotated independently by multiple curators based on the observed spatial expression pattern. All mouse work was approved by the University of California–San Francisco Institutional Animal Care and Use Committee.
Results
Clinical report
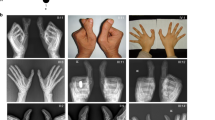
All affected family members had a phenotype that corresponds to triphalangeal thumb–polysyndactyly syndrome (TPT-PS, OMIM 174500). The pedigree demonstrated an autosomal dominant inheritance of the phenotype. All patients were bilaterally affected. They presented with at least one triphalangeal thumb on both hands. The hands typically demonstrated a poly- and syndactylous block of digits on the anterior and posterior side, with the second and occasionally the third digit present between both blocks. Additionally, postaxial syndactyly and polydactyly of both feet were observed in patients V-4, V-5, and V-6. The affected family members did not present with other congenital anomalies. (Figure 2 and Supplementary Figure S3)

Clinical images of triphalangeal thumb–polysyndactyly syndrome (TPT-PS) phenotypes of patients V-5 and V-6. The full color image of this figure is available in the online version of the paper at http://www.nature.com/gim.
TPT-PS is linked to 7q36
Genome-wide linkage analysis was performed to validate the linkage results that were obtained by Tsukurov et al.15 With SNP-array data of all 10 included family members, LOD scores were calculated using easyLINKAGE software. Linkage was confirmed at chromosome 7q36 (LOD score 3.0, chr7:156033299-158113390, GRCh37/hg19). This region corresponds to the region that was found in the same TPT-PS family by Tsukurov et al.15 (LOD score D7S550). Additionally, a maximum LOD score of 3.0 was found on chromosome 21q22.11 (chr21:33405700-36673573, GRCh37/hg19).
Absence of ZRS and copy-number disease-associated variations
As the ZRS has been associated with various limb malformations, we analyzed our family for mutations and CNVs in this region. The ZRS was sequenced in patients V-4, V-5, and V-6 and did not reveal any disease-associated alterations. Because TPT-PS is commonly associated with duplications including the ZRS,10 SNP-array data of all TPT-PS patients were reviewed to identify CNVs on chromosome 7q36. No CNVs larger than 2.1 kb were found on chromosome 7q36 (Figure 3) or chromosome 21q22.11. Finally, multiplex ligation-dependent probe amplification analysis did not reveal pathogenic deletions or duplications between exon 3 and exon 6 of LMBR1.

Fluorescence in situ hybridization and copy-number variations analysis. (a) Results of fluorescence in situ hybridization (FISH) analysis of patient V-5. The location of five FISH probes are depicted in the genome track. The labeled colors of each probe correspond to the color displayed on the images. (b) Images of Nexus copy-number analysis (Illumina Infinium GSAMD-24v1 microarray) in patients V-5 and V-6 do not show copy-number variations on chromosome 7q36. pZRS, pre–zone of polarizing activity regulatory sequence; ZRS, zone of polarizing activity regulatory sequence. The full color image of this figure is available in the online version of the paper at http://www.nature.com/gim.
Karyotyping using GTC-banded chromosomes revealed no aberrant chromosomal pattern in the two tested individuals. FISH with dual-labeled BAC probes using different probe combinations (Figure 3, Supplementary Figure S4 online) excluded small subchromosomal rearrangements in the SHH-LMBR1 region or the telomeric part of chromosome 7q36. Also, using a BAC probe covering the ZRS region in intron 5 of the LMBR1 gene (RP11-51I24) we did not detect any aberrant patterns of hybridization in interphase nuclei or banded chromosomes.
No pathogenic exonic variants linked to limb defects
We next wanted to test whether coding mutations in other genes might be associated with this phenotype. Samples of four affected patients were investigated with whole-exome sequencing to verify if an exonic pathogenic variant was present on chromosome 7q36, 21q22.11 or in other coding regions. The affected family members did not share any predicted pathogenic variants with an allele frequency of less than 0.001 in 1000 Genomes,20 ExAC,21 and Human Gene Mutation Database22 data sets.
Identification of a point mutation in the pZRS
After sequencing a 4.5-kb region covering the ZRS and pZRS, a heterozygous G>C point mutation in the pZRS (chr7:156585476G>C; hg19) was identified (Figure 4). This point mutation was found in all affected patients and was absent in an unaffected family member. The variant was not present in online databases (dbSNP,23 ClinVar,24 ExAC,21 and Human Gene Mutation Database) or in locally available whole-genome sequencing data sets (GoNL25 and Wellderly26).

Mutation location of the identified point mutation in intron 5 of LMBR1. The pre–zone of polarizing activity regulatory sequence (pZRS) is visualized through the conservation track in the University of California–Santa Cruz Genome Browser. The chromatogram of three affected family members reveal a G>C point mutation. The full color image of this figure is available in the online version of the paper at http://www.nature.com/gim.
Computation analyses of pZRS variants
Previous reported mutations in the pZRS were found to only lead to preaxial polydactyly in dogs12 and humans.13 However, our identified pZRS mutation, chr7:156585476G>C, leads to a much more severe phenotype, TPT-PS, which also includes the lower limbs. To test for potential causes for these phenotypic differences, we carried out various computational analyses on these different mutations. Evolutionary conservation analyses of this variant compared with the previously discovered canine and human variants using both PhyloP and genomic evolutionary rate profiling27 did not find chr7:156585476G to have higher conservation scores than these previously reported variants (Supplementary Figure S5 online). Additionally, Combined Annotation Dependent Depletion scores28 for this nucleotide were not higher than these other variants (Supplementary Figure S5 online). Combined, these analyses could not explain the cause for the stronger limb phenotype observed in this family.
Furthermore, TPT-PS is widely associated with genomic duplications that cover the ZRS. Duplications that include the ZRS are also able to cause more severe anomalies, like Haas-type polysyndactyly and Laurin–Sandrow syndrome.10 The similarity of phenotypes in pZRS mutations and genomic duplications indicate corresponding levels and patterns of SHH disruption in the limb bud. This similarity also introduces the hypothesis that SHH regulation in pZRS mutations and genomic duplications might be affected by the same underlying molecular mechanism.
Although genomic duplications encompassing the ZRS have regularly been reported in the literature, no study has investigated the pathogenic mechanism that ultimately results in these severe limb anomalies. Two hypotheses have been suggested regarding genomic duplications of the ZRS. First, it has been proposed that a genomic duplication could have a dosage effect, affecting the regulatory balance between enhancers and promoters.29
Second, genomic duplications are able to alter the chromatin organization of the entire genome.30 Hi-C sequencing has revealed that the chromatin structure is partitioned into various TADs. Duplications of the ZRS therefore might disrupt a TAD boundary in the vicinity of the ZRS and cause limb anomalies through one of these mechanisms. Through the Hi-C 3D-genome browser and ENCODE track for chromatin organization, we mapped the predicted TAD boundaries and CTCF and RAD21 binding sites in the SHH-ZRS domain31,32 (Supplementary Figure S6 online). The Hi-C genome browser and the presence of increased CTCF and RAD21 binding sites predicted a TAD boundary in intron 4 of LMBR1, 25 kb upstream of the ZRS. Lohan et al.10 reported 16 different sizes of genomic duplications encompassing the ZRS that caused severe limb anomalies. Fifteen of these duplications also include the predicted TAD boundary in intron 4 of LMBR1. However, the smallest duplication of 16 kb in a Laurin–Sandrow syndrome family did not surpass this TAD boundary. As the phenotypes of genomic duplications and point mutations in the pZRS have close similarities, we assessed if the pZRS represents an additional TAD boundary that can be disrupted by point mutations. We did not identify increasing CTCF and RAD21 binding sites in the pZRS region (Supplementary Figure S6 online).
Therefore, although the disruption of the TAD boundary in the vicinity of the ZRS complex could explain the pathogenic mechanism in TPT-PS families with genomic duplications, it is not able to elucidate the mechanism in the family with a point mutation in the pZRS or the previously reported family with the 16-kb duplication.
chr7:156585476G change leads to ectopic ZRS expression in the developing limb bud
We next carried out mouse transgenic enhancer assays on both the reference and mutant allele to test whether it leads to any altered enhancer activity. Both the pZRS and ZRS region for each allele were cloned into a mouse enhancer assay vector and analyzed for their LacZ expression at E11.5 in transgenic mice. For the reference allele, eight transgenic assays were made, of which seven showed expression in the posterior margin of the limb bud (Figure 5, Supplementary Figure S7 online). We observed a significant expansion of LacZ expression for the TPT-PS associated allele, extending to the entire mesenchyme all the way into the trunk region in five of six mutated transgenic enhancer assays made (Figure 5, Supplementary Figure S7 online). In addition, the mutant allele also showed LacZ expression in the mandibular process that was not observed in the reference sequence. It is worth noting that both alleles also showed consistent LacZ staining in the snout, similar to that previously observed for the canine sequence,12 and that the reference allele showed slight expression in the anterior autopod (Figure 5, Supplementary Figure S7 online) different from previous studies that tested ZRS only33 or the canine ZRS + pZRS in mice.12 In sum, our results show that transgenic mice carrying the mutated pZRS allele have increased enhancer activity dispersed throughout the entire limb bud including the trunk region at the base of the limb compared with the reference allele.

LacZ expression in mice embryos (E11.5) carrying the (a) wild-type zone of polarizing activity regulatory sequence (ZRS)/pre-ZRS (pZRS) construct or the (b) mutated ZRS/pZRS construct. The full color image of this figure is available in the online version of the paper at http://www.nature.com/gim.
Discussion
In this study, we report a Dutch family with triphalangeal thumb–polysyndactyly syndrome with a point mutation in a conserved noncoding regulatory region in the genome; the pZRS. This point mutation was identified after several abnormalities known to cause a TPT-PS phenotype were ruled out, including point mutations, CNVs, and chromosomal rearrangements of the ZRS as well as pathogenic variants in other genes. We discovered a novel mutation in the pZRS that segregated with the disease and showed differential enhancer activity in transgenic mice.
Previously reported pZRS mutations
Two studies have previously associated point mutations in the pZRS with congenital limb anomalies. Park et al.12 identified two point mutations in the pZRS in two different breeds of dogs, but did not find altered expression in an enhancer assay. However, it is important to also note that this mutation is located near the telomeric end of the pZRS (Supplementary Figure S5 online). In another study, Xiang et al.13 screened a large population of children with nonsyndromic preaxial polydactyly for mutations in SHH, GLI3, ZRS, and pZRS. Patients with isolated preaxial polydactyly of the thumb are often unilaterally affected and do not have a positive family history. Therefore, it is questionable whether this phenotype is due to underlying genetic aberrations. In addition, these mutations were not followed up via an enhancer assay for their functional consequences. Computational analyses of our identified mutation did not show increased conservation scores, which could have potentially explained the more severe phenotype.
The role of the pZRS in this SHH-ZRS complex
The functional role of the pZRS remains largely unknown. The pZRS was delineated from the ZRS by Park et al.12 and is well conserved among vertebrate species and positioned approximately 750 bp 5’ upstream of the ZRS. Until the present study, there was no functional or molecular evidence confirming that the pZRS is an independently functioning regulatory element. Considering the close proximity of the pZRS and the ZRS, the pZRS could potentially function as an independent regulatory element of SHH but also could function alongside the ZRS.
The central part of the ZRS has previously been shown to have a role in regulating the levels of SHH expression in the limb,34 with the 5’ end involved in spatiotemporal functions and the 3’ end required for long-range activity of the ZRS that ensures SHH signaling.34,35 Additionally, different ETV sites throughout the ZRS govern the posterior restriction of SHH to the ZPA.36 Further molecular and functional studies will be required to determine which regulatory role the pZRS has and how it is associated with SHH limb expression.
Genotype–phenotype correlation
The observation of severe TPT phenotypes in combination with a single pZRS point mutation in this family provides insights into the possible underlying pathogenic mechanism of this region. The TPT-PS phenotype is clearly more severe than TPT phenotypes caused by previously described point mutations in the ZRS. Several studies have shown that point mutations in the ZRS affect the activity of the ZRS by inserting or removing transcription factor binding sites of ETS, ETV, HOX, and TFAP2B.34,36,37 These point mutations generally lead to isolated TPT or TPT with one additional thumb. Other described point mutations in the ZRS, however, do cause more severe anomalies than isolated TPT with polydactyly, such as those involving positions 402 and 404 that are associated with isolated TPT with tibial hypoplasia.6,38,39 In contrast to our reported family, the severe anomalies in 402 and 404 point mutations are only observed in the lower limb and not in the upper limb.
A recent study also revealed that a point mutation in position 105 causes severe TPT phenotypes that resemble TPT-PS in several affected family members of a large Dutch TPT population.40 However, the simultaneous presence of less severe phenotypes such as isolated TPT and TPT with preaxial polydactyly in other family members indicate that additional nongenetic factors or genetic modifiers are required to magnify SHH disruption in the limb bud. Considering the severe phenotype in the present Dutch family, it is unlikely that alterations in transcription factor binding site affinity have the ability to induce the level of disruption that is required to cause consistent TPT-PS phenotypes in this family. Furthermore, as TPT-PS is mainly observed in families with genomic duplications that encompass the ZRS, we evaluated whether pZRS mutations can alter the integrity of TAD boundaries in the SHH-LMBR1 topological domain. We did not identify increased presence of CTCF or RAD21 binding sites in the pZRS. Additionally, the TAD boundary seems to be located approximately 25 kb from the telomeric end of the pZRS (Supplementary Figure S6 online). Therefore, pZRS mutations are not likely to be involved in the disruption of the SHH-LMBR1 TAD.
Altered enhancer activity
Our mouse enhancer assay that encompassed both the ZRS and pZRS showed enhancer expression beyond the normal posterior ZPA expression pattern of previous transgenic mice carrying the human ZRS sequence33 or the dog ZRS + pZRS sequence.12 For the dog mutations, similar mouse transgenic enhancer assays did show a slight expansion of enhancer activity in the posterior limbs compared with the reference sequence.12 In our study, mice carrying the reference allele did show slight anterior expression, but those carrying the mutated allele showed significantly more extended enhancer expression. This extended ectopic expression pattern could explain the severity of the limb phenotype in this family and suggests that the pZRS is involved in SHH expression and limb embryonic development and that mutations in this region can cause a severe triphalangeal thumb–polysyndactyly phenotype.
The regulatory network of SHH remains to be completely elucidated. New initiatives combining information from the molecular, genetic, and clinical points of view will help to elucidate the complex regulatory network in this locus that may also serve as a model for understanding long-range regulatory mechanisms for other loci.
References
Dixon JR, Selvaraj S, Yue F et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012;485:376–80.
Williamson I, Lettice LA, Hill RE & Bickmore WA. Shh and ZRS enhancer colocalisation is specific to the zone of polarising activity. Development 2016;143:2994–3001.
Jeong Y, El-Jaick K, Roessler E, Muenke M & Epstein DJ. A functional screen for sonic hedgehog regulatory elements across a 1 Mb interval identifies long-range ventral forebrain enhancers. Development 2006;133:761–72.
Muller F, Chang B, Albert S, Fischer N, Tora L & Strahle U. Intronic enhancers control expression of zebrafish sonic hedgehog in floor plate and notochord. Development 1999;126:2103–16.
Hill RE, Heaney SJ & Lettice LA. Sonic hedgehog: restricted expression and limb dysmorphologies. J Anat 2003;202:13–20.
Lettice LA, Heaney SJ, Purdie LA et al. A long-range Shh enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly. Hum Mol Genet 2003;12:1725–35.
Lettice LA, Hill AE, Devenney PS & Hill RE. Point mutations in a distant sonic hedgehog cis-regulator generate a variable regulatory output responsible for preaxial polydactyly. Hum Mol Genet 2008;17:978–85.
Maas SA, Suzuki T & Fallon JF. Identification of spontaneous mutations within the long-range limb-specific Sonic hedgehog enhancer (ZRS) that alter Sonic hedgehog expression in the chicken limb mutants oligozeugodactyly and silkie breed. Dev Dyn 2011;240:1212–22.
VanderMeer JE & Ahituv N. Cis-regulatory mutations are a genetic cause of human limb malformations. Dev Dyn 2011;240:920–30.
Lohan S, Spielmann M, Doelken SC et al. Microduplications encompassing the Sonic hedgehog limb enhancer ZRS are associated with Haas-type polysyndactyly and Laurin-Sandrow syndrome. Clin Genet 2014;86:318–25.
Petit F, Jourdain AS, Holder-Espinasse M et al. The disruption of a novel limb cis-regulatory element of SHH is associated with autosomal dominant preaxial polydactyly-hypertrichosis. Eur J Hum Genet 2016;24:37–43.
Park K, Kang J, Subedi KP, Ha JH & Park C. Canine polydactyl mutations with heterogeneous origin in the conserved intronic sequence of LMBR1. Genetics 2008;179:2163–72.
Xiang Y, Jiang L, Wang B, Xu Y, Cai H & Fu Q. Mutational screening of GLI3, SHH, preZRS, and ZRS in 102 Chinese children with nonsyndromic polydactyly. Dev Dyn 2017;246:392–402.
Nicolai JP & Hamel BC. A family with complex bilateral polysyndacty. J Hand Surg Am 1988;13:405–7.
Tsukurov O, Boehmer A, Flynn J et al. A complex bilateral polysyndactyly disease locus maps to chromosome 7q36. Nat Genet 1994;6:282–6.
Thiele H & Nurnberg P. HaploPainter: a tool for drawing pedigrees with complex haplotypes. Bioinformatics 2005;21:1730–2.
McGowan-Jordan J, Simons A, Schmid M(eds). An International System for Human Cytogenomic Nomeclature. Karger: Basel, Switzerland, 2016.
Eussen BH, van de Laar I, Douben H et al. A familial inverted duplication 2q33-q34 identified and delineated by multiple cytogenetic techniques. Eur J Med Genet 2007;50:112–9.
Wang K, Li M & Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010;38:e164.
The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 2015;526:68–74.
Lek M, Karczewski KJ, Minikel EV et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016;536:285–91.
Stenson PD, Mort M, Ball EV et al. The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet 2017;136:665–77.
Sherry ST, Ward MH, Kholodov M et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 2001;29:308–11.
Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res 2016;44:D862–8.
Genome of the Netherlands Consortium. Whole-genome sequence variation, population structure and demographic history of the Dutch population. Nat Genet 2014;46:818–25.
Erikson GA, Bodian DL, Rueda M et al. Whole-genome sequencing of a healthy aging cohort. Cell 2016;165:1002–11.
Cooper GM, Stone EA, Asimenos G et al. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res 2005;15:901–13.
Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM & Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014;46:310–5.
Will AJ, Cova G, Osterwalder M et al. Composition and dosage of a multipartite enhancer cluster control developmental expression of Ihh (Indian hedgehog). Nat Genet 2017;49:1539–45.
Franke M, Ibrahim DM, Andrey G et al. Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature 2016;538:265–9.
Rosenbloom KR, Sloan CA, Malladi VS et al. ENCODE data in the UCSC Genome Browser: year 5 update. Nucleic Acids Res 2013;41(Database issue):D56–63.
Wang Y, Zhang B, Zhang L et al. The 3D Genome Browser: a web-based browser for visualizing 3D genome organization and long-range chromatin interactions[preprint]. bioRxiv 2017: 112268.
Laurell T, Vandermeer JE, Wenger AM et al. A novel 13 base pair insertion in the sonic hedgehog ZRS limb enhancer (ZRS/LMBR1) causes preaxial polydactyly with triphalangeal thumb. Hum Mutat 2012;33:1063–6.
Lettice LA, Devenney P, De Angelis C & Hill RE. The conserved sonic hedgehog limb enhancer consists of discrete functional elements that regulate precise spatial expression. Cell Rep 2017;20:1396–408.
Lettice LA, Williamson I, Devenney PS, Kilanowski F, Dorin J & Hill RE. Development of five digits is controlled by a bipartite long-range cis-regulator. Development 2014;141:1715–25.
Lettice LA, Williamson I, Wiltshire JH et al. Opposing functions of the ETS factor family define Shh spatial expression in limb buds and underlie polydactyly. Dev Cell 2012;22:459–67.
Fuxman Bass JI, Sahni N, Shrestha S et al. Human gene-centered transcription factor networks for enhancers and disease variants. Cell 2015;161:661–73.
Girisha KM, Bidchol AM, Kamath PS et al. A novel mutation (g.106737G>T) in zone of polarizing activity regulatory sequence (ZRS) causes variable limb phenotypes in Werner mesomelia. Am J Med Genet A 2014;164A:898–906.
VanderMeer JE, Lozano R, Sun M et al. A novel ZRS mutation leads to preaxial polydactyly type 2 in a heterozygous form and Werner mesomelic syndrome in a homozygous form. Hum Mutat 2014;35:945–8.
Baas M, Potuijt JWP, Hovius SER, Hoogeboom AJM, Galjaard RH & van Nieuwenhoven CA. Intrafamilial variability of the triphalangeal thumb phenotype in a Dutch population: evidence for phenotypic progression over generations? Am J Med Genet A 2017;173:2898–2905.
Acknowledgments
We thank Jim McGill for the genetic counseling of Australian family members and the staff of the Histopathological and Molecular Pathology Department at the Mater Hospital, Brisbane, Australia, with special thanks to Gareth Price for incremental help on the whole-exome-sequencing analysis.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Potuijt, J.W.P., Baas, M., Sukenik-Halevy, R. et al. A point mutation in the pre-ZRS disrupts sonic hedgehog expression in the limb bud and results in triphalangeal thumb–polysyndactyly syndrome. Genet Med 20, 1405–1413 (2018). https://doi.org/10.1038/gim.2018.18
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2018.18
Keywords
This article is cited by
-
Genetic determinants of syndactyly: perspectives on pathogenesis and diagnosis
Orphanet Journal of Rare Diseases (2022)
-
Evolutionary genetics of flipper forelimb and hindlimb loss from limb development-related genes in cetaceans
BMC Genomics (2022)
-
The pZRS non-coding regulatory mutation resulting in triphalangeal thumb–polysyndactyly syndrome changes the pattern of local interactions
Molecular Genetics and Genomics (2022)
-
A 300-kb microduplication of 7q36.3 in a patient with triphalangeal thumb-polysyndactyly syndrome combined with congenital heart disease and optic disc coloboma: a case report
BMC Medical Genomics (2020)
