
Abstract
Purpose
This study aimed to identify the genetic cause of a new multiple congenital anomalies syndrome observed in three individuals from two unrelated families.
Methods
Clinical assessment was conducted prenatally and at different postnatal stages. Genetic studies included exome sequencing (ES) combined with single-nucleotide polymorphism (SNP) array based homozygosity mapping and trio ES. Dermal fibroblasts were used for functional assays.
Results
A clinically recognizable syndrome characterized by severe developmental delay, variable brain anomalies, congenital heart defects, dysmorphic facial features, and a distinctive type of synpolydactyly with an additional hypoplastic digit between the fourth and fifth digits of hands and/or feet was identified. Additional features included eye abnormalities, hearing impairment, and electroencephalogram anomalies. ES detected different homozygous truncating variants in MAPKAPK5 in both families. Patient-derived cells showed no expression of MAPKAPK5 protein isoforms and reduced levels of the MAPKAPK5-interacting protein ERK3. F-actin recovery after latrunculin B treatment was found to be less efficient in patient-derived fibroblasts than in control cells, supporting a role of MAPKAPK5 in F-actin polymerization.
Conclusion
Our data indicate that loss-of-function variants in MAPKAPK5 result in a severe developmental disorder and reveal a major role of this gene in human brain, heart, and limb development.
Similar content being viewed by others

INTRODUCTION
The mitogen-activated protein kinase (MAPK) signaling pathways mediate the reaction of the cell to external stimuli by regulating fundamental physiological processes such as cell proliferation, differentiation, apoptosis, survival, gene expression, and cell motility.1,2
Mammalian cells have been described with four well-characterized canonical MAPK pathways, each consisting of three distinct protein kinase units: a MAPK, a MAPK kinase (MAPKK), and a MAPK kinase kinase (MAPKKK). Signaling mediated by MAPKs originates on the cell surface upon external stimulation and propagates from one kinase to the next through a consecutive series of protein phosphorylation and subsequent activation events. Typical extracellular inducers of MAPK cascades are environmental stresses, growth factors, hormones, cytokines, or mitogens. MAPKs are at the base of the signaling cascades and operate by phosphorylating a broad range of substrates including downstream MAPK-activated protein kinases (MAPKAPK), phosphatases, transcription factors, and other proteins to build the corresponding cellular response to the initial stimulus.1,2
MAPK activated protein kinase 5 (MAPKAPK5; MIM 606723), also designated as p38-regulated and-activated kinase (PRAK), or MK5, is a serine/threonine protein kinase first identified as a MAPKAPK2 (MK2, MIM 602006) homolog by database similarity search (MK2-MK5 amino acid identity: 30% by NCBI BLAST sequence alignment).3 The gene encoding MK5 (MAPKAPK5) lies on chromosome 12q24.12-q24.13, and consists of 14 coding exons that lead to protein isoforms of 471 and 473 residues by alternative splicing (GeneBank: NM_003668.3, NM_139078.2). MK5 has been reported to mainly localize to the nucleus and be activated by the canonical MAPK p38MAPK,3,4,5,6 and the atypical MAPKs ERK3 (MAPK6) and ERK4 (MAPK4);7,8,9,10 the last two kinases having strong physiological interaction with MK5.7,9,10 ERK3 and ERK4 differ from conventional MAPKs in the sequence of their activation loop (Ser-Glu-Gly instead of Thr-Xxx-Tyr), and also have a distinctive C-terminal domain.2 Although so far it is unclear which extracellular stimuli lead to ERK3/4 activation, both proteins are phosphorylated by group I p21-activated kinases (PAKs 1-3), which regulate transcription, apoptosis, cell proliferation, and cell motility.11
We report the identification of biallelic truncating variants in MAPKAPK5 in three affected individuals from two unrelated families with a novel syndromic form of neurocardiofaciodigital congenital anomalies. Individuals having two MAPKAPK5 truncating alleles shared a common phenotype characterized by severe global developmental delay, congenital heart defects, brain anomalies, a distinctive type of postaxial synpolydactyly, and facial dysmorphic features.
MATERIALS AND METHODS
Molecular genetic analysis
Genome wide single-nucleotide polymorphism (SNP) array hybridization using CytoSNP-850K Beadchip arrays (Illumina, San Diego, CA) and exome sequencing (ES) (Sistemas Genomicos, Valencia, Spain) in individuals from family 1 were performed using peripheral blood genomic DNA according to previously described protocols.12 ES reads were aligned to human genome assembly (GRCh37/hg19) and variants were filtered by minor allele frequency (MAF) with respect to population databases (absent or <0.01 in 1000 Genomes) and by functional prediction (missense, nonsense, indel, or splice site variants). For family 2, genomic DNA from affected individual 3 and his parents was isolated from peripheral blood and enriched with a SureSelect Human All Exon Kit V6 (Agilent technologies, Santa Clara, CA) for subsequent trio ES on an Illumina system (Illumina, San Diego, CA). Reads were aligned to human genome build GRCh37/hg19. Sequence reads were called and analyzed using the VarFish platform13 and variant filtration was applied as previously published.14
Dermal fibroblasts
Primary fibroblasts from affected individual 1 were developed from a skin biopsy following methods described earlier.12 Neonatal and adult control human dermal fibroblasts were obtained commercially as follows: C1 (Lonza, Basel, Switzerland, cc2509 [neonatal]); C2 (Sigma-Aldrich, St Louis, MO, 106-05N [neonatal]); C3 (Lonza, cc2511 [adult]). All cells were tested negative for mycoplasma.
Quantitative real-time reverse transcription PCR (qRT-PCR)
Total RNA from cultured fibroblasts was isolated using Tri Reagent solution (Invitrogen, Thermo Fisher Scientific, Waltham, MA) following the manufacturer´s protocol and retrotranscribed with the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Thermo Fisher Scientific). qRT-PCR analysis was performed using TaqMan real-time PCR gene expression assays (Applied Biosystems) in a 7900HT Fast Real-Time PCR System (Applied Biosystems). For each line of fibroblasts, RNA from three independent extractions was analyzed and every sample was run in triplicates. RNA levels were referred to housekeeping genes GUSB or GAPDH. Fold differences in gene expression were calculated by the 2−ΔΔCt method using the mean value of ΔCt from C1 as the calibrator sample. Taqman assays: Hs00177572_m1 and Hs00934300_m1 for MAPKAPK5, Hs99999905_m1 for GAPDH and Hs99999908_m1 for GUSB.
Immunoblotting
Human primary fibroblasts seeded onto p100 plates were incubated until confluence at 37 °C, 5% CO2 in growth medium (Dulbecco’s Modified Eagle’s Medium [DMEM] + 10% FBS + 1% AA [antibiotic/antimycotic], [Gibco, Thermo Fisher Scientific]). Subsequently, cells were washed in cold PBS and lysed in RIPA buffer (150 mM NaCl, 50 mM TrisHCl, 2 mM EDTA, 0.5% sodium deoxicolate, 0.1% SDS, 1% NP-40) supplemented with protease inhibitors (Sigma-Aldrich, St. Louis, MO, P8340, 1 mM PMSF) and phosphatase inhibitors (Sigma-Aldrich, P0044 and P5726, 1 mM Na3O4V and 10 mM NaF). Cell lysates were incubated 20 minutes on ice with vortexing every 5 minutes and clarified by centrifugation at 4 °C for 15 minutes at 14,000g. Protein concentration was determined by the BCA colorimetric method (Pierce, Thermo Fisher Scientific). A total amount of 30 μg of protein were separated on 8% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gels and transferred onto nitrocellulose membranes (GE Healthcare, Buckinghamshire, UK). The top part of blots was incubated with anti-ERK3 (Cell Signaling Technology, Leiden, The Netherlands, #4067; 1:1,000) and the lower part with an anti-MK5 antibody developed against a synthetic peptide corresponding to residues surrounding Gly367 (Cell Signaling Technology, D70A10, #7419; 1:1000). Tubulin (TUB; Sigma-Aldrich, T9026; 1:80,000) was evaluated on the anti-MK5 blot after 30 minutes incubation with 0.06% azide-TTBS. HRP-conjugated secondary antibodies (1:10,000) were from Jackson ImmunoResearch (Cambridgeshire, UK). Immunoblots were developed using ECL reagent (GE Healthcare, Chalfont St. Giles, UK) and exposed on Agfa X-ray films. Densitometric analysis was performed with the help of ImageJ software.
Remodeling F-actin cytoskeleton assay
Filamentous-actin (F-actin) recovery assay was conducted following the protocol described by Kosmas et al.15 Briefly, fibroblasts were seeded on coverslips in 24-well plates (2 × 104 cells/well) in growth medium and incubated the next day for 30 minutes in DMEM containing 2.5 µM latrunculin B (Abcam, Cambridge, UK, ab144291) previously dissolved in DMSO. Subsequently, the media was removed and the cells were washed three times with PBS to eliminate any traces of latrunculin B. Fibroblasts were left to recover in growth medium before being fixed at different time points with 4% PFA/PBS 10 minutes on ice. Following permeabilization in 0.1% Triton X-100/PBS 10 minutes, cells were stained for 30 minutes with Phalloidin-546 (Molecular Probes, Thermo Fisher Scientific, A22283, 1:200) and DAPI (Molecular Probes, 1:1500) in 0.05% Tween/PBS to visualize F-actin filaments and nuclei, respectively. Images were acquired on a Nikon 90i microscope.
F-actin/G-actin fluorescence quantification
For cytofluorescence evaluation of F-actin versus free globular-monomeric-actin (G-actin), fibroblasts were seeded on coverslips in growth medium (3.5 × 104 cells/well [24-well plate]), treated with latrunculin B/DMSO (2.5 µM) or DMSO as above, washed in PBS, and either fixed in 4% PFA/PBS or cultured (latrunculin B-treated) in growth medium for an additional 30 minutes before fixation. In each experimental replicate, untreated cells used as reference were simultaneously seeded at the same density, maintained in DMEM for 30 minutes, washed in PBS, and cultured in growth medium for 30 minutes before fixation. Fixed cells were permeabilized as indicated above and incubated with Alexa Fluor 488 phalloidin (1:500, Molecular probes A12379) and Alexa Fluor 594 Deoxyribonuclease I (DNase I; 1:500, Molecular Probes D12372) for labeling of F-actin and G-actin respectively, and DAPI for 30 minutes. Subsequently, cells were washed in 0.05% Tween-20/PBS followed by two times in PBS and mounted with Prolong Diamond (Molecular Probes) for confocal microscopy (Zeiss LSM710). For each line of fibroblasts, three images (Z-project) per condition were acquired in every experimental replicate and fluorescence quantification was determined in individual cells with the help of ImageJ Fiji software. Analyzed cells were delimited with a ROI and the integrated density value for Phalloidin and DNase I in each cell was calculated. A threshold for each fluorescence channel was used to exclude nonstained areas. This threshold was maintained constant within the same experimental replicate in control and patient fibroblasts.
Statistical analyses
Statistical analysis was conducted with GraphPad 6.0 software. Normality and homoscedasticity of the samples was checked with Shapiro–Wilk, Kolmogorov–Smirnov normality tests or Skewness and Kurtosis normality score, and Brown–Forsythe or Bartlett´s variance F-test respectively. When the data met both requirements, one-way analysis of variance (ANOVA) with Tukey´s multiple comparison test was performed. Otherwise, nonparametric two-tailed Mann–Whitney test was used. No randomization, predetermination of sample size nor blinding were used.
RESULTS
Identification of a new type of developmental disorder
The proband of family 1, affected individual 1, is a girl born to an Egyptian first cousin couple with a family history of one deceased child, one previous infant death and a termination of pregnancy (see case reports in Supplemental data). Fetal ultrasound examination of affected individual 1 indicated intrauterine growth retardation and tetralogy of Fallot (TF). At 40 weeks of gestation, her birth length and occipitofrontal head circumference (OFC) were normal, and birth weight was reduced. After birth, the heart defect was surgically corrected. Facial dysmorphisms including broad forehead, sparse hair, narrow palpebral fissures, thin lips, and retromicrognathia were observed (Fig. 1a). Limb examination revealed synpolydactyly of hands with bilateral double nail of the fifth fingers and partial syndactyly between fourth and fifth fingers (Fig. 1e, f). Her feet also showed synpolydactyly with a short extra digit between the fourth and fifth toes on the right foot and broad short left fifth toe with double nail as well as syndactyly between fourth and fifth toes on the left foot (Fig. 1h). Radiological examination of hands and feet revealed duplication of middle and terminal phalanges of the right fifth finger and duplication of the terminal phalanx of left fifth finger as well as an extra hypoplastic metatarsal and extra phalanx between the fourth and fifth toes (Fig. 1g, i).

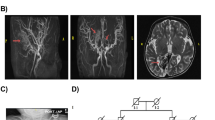
(a) Face of affected individual 1 at 9 years showing bitemporal narrowing, long face, overhanging nasal tip, and thin lips. (b) Face of the younger sister, affected individual 2, at 2 years of age showing bitemporal narrowing, prominent forehead, sparse eyebrows, prominent nasal tip, wide nares, thin lips, and microretrognathia. (c–d) Affected individual 3 at age of 19 months with short and narrow palpebral fissures, a small mouth with thin lips, and uplifted ear lobes (c). Retrognathia and dysmorphic ears with missing crus of superior antihelix and nearly missing ear lobe can be observed in (d). (e–f). Hands of affected individual 1 showing synpolydactyly: bilateral double nail of the little fingers with partial cutaneous syndactyly between the fourth and duplicated fingers in both hands. (g) X-ray of hands of affected individual 1 showing duplication of terminal phalanx of left little finger and duplication of middle and terminal phalanges of the right little finger with short first metacarpals bilaterally. (h) Image of the right foot of affected individual 1 showing deep groove between first and second toes and a short extra digit between the fourth and fifth toes. (i) X-ray (affected individual 1) showing an extra hypoplastic metatarsal bone and an extra phalanx between the fourth and fifth toes, absent middle and terminal phalanges of the fifth toe and hypoplastic middle and terminal phalanges of the second, third, and fourth toes in right foot. (j) Affected individual 3: synpolydactyly with an additional hypoplastic toe between the fourth and fifth toes of the right foot. (k) X-ray of the right foot of affected individual 3 showing synpolydactyly with an extra hypoplastic metatarsal bone between the fourth and fifth metatarsals and an extra proximal phalangeal bone between the fourth and fifth toes. (l–m) Magnetic resonance image (MRI) brain of affected individual 1 at 9 years. L: T2 axial cut showing mild demyelination around the occipital horn of the lateral ventricles. M: sagittal cut showing thin body of corpus callosum, prominent cerebellar folia, and vermian hypoplasia. The fourth ventricle is communicating with extra axial cerebrospinal fluid (CSF). (n,o). MRI brain of affected individual 2 at 18 months. (n) T2 axial cut showing cavum septum pellucidum, asymmetric dilatation of lateral ventricles with more dilatation on the left side. (o) Sagittal cut showing thin arched corpus callosum, prominent cerebellar folia, and mild vermian hypoplasia. The fourth ventricle is communicating with extra axial CSF.
Brain anomalies detected by repeated magnetic resonance imaging (MRI) and computer tomography (CT) studies showed dilated fourth ventricles communicating with extra axial cerebrospinal fluid (CSF), foramen of Luschka and cisterna magna dilatation, periventricular demyelination of the lateral ventricles, and hypoplasia of the cerebellar vermis (Fig. 1l, m). Electroencephalogram (EEG) showed abnormal findings consistent with generalized epileptogenic discharge of left side origin.
Ophthalmologic examination revealed pallor of optic discs and tessellated fundus of the retina. Visual evoked potentials (VEP) showed mild post visual pathway dysfunction. Moderate loss of hearing was detected by auditory brainstem response (ABR).
At the age of 9 years she had severe global developmental delay; she was unable to stand unsupported and only said one word. Anthropometric measurements at this age revealed failure to thrive (weight: -7 SD), short stature (height: -3.5 SD), and normal OFC.
The family had another child, affected individual 2, who was born preterm at 36 weeks of gestation with a normal birth weight. Dysmorphic features included bitemporal narrowing, prominent forehead, sparse eyebrows, narrow palpebral fissures, microretrognathia, low-set simple ears, and overlapping fingers with hypoplastic nails (Fig. 1b). Echocardiography showed patent ductus arteriosus (PDA) and atrial septal defect (ASD). MRI brain scan at the age of 18 months revealed cavum of septum pellucidum, dilatation of lateral ventricles, thin arched corpus callosum, prominent cerebellar folia, mild vermian hypoplasia, and fourth ventricle communicating with extra axial CSF (Fig. 1n, o). Brain CT at 1.5 years of age showed dilated supra- and infratentorial ventricles with no CSF permeation and cortical sulci effacement suggestive of hydrocephalus.
EEG showed generalized abnormalities and seizures in this individual were controlled by phenobarbital. Abdominopelvic ultrasound showed right hydroureteronephrosis. Micturating cystourethrogram revealed evidence of significant vesicoureteral reflux grade V. Ophthalmological assessment was normal apart from nystagmus.
At the age of 2 years and 7 months the affected child was severely delayed with no head control and no speech development. Anthropometric measurements at 3 years of age showed failure to thrive (weight: -2.4 SD), short stature (height: -3.8 SD) and microcephaly (OFC: -2.7 SD).
The proband of family 2, affected individual 3, is the first child of healthy consanguineous parents (first cousins) of Turkish descent. A hypoplastic left heart was diagnosed by prenatal ultrasound examination. He was delivered in the 38th week of pregnancy by cesarean section (CS) with normal measurements. Pierre-Robin sequence (retrognathia, glossoptosis, high arched palate without cleft), short, narrow, and upslanted palpebral fissures, a small mouth with thin lips, uplifted ear lobes, a short neck, and nystagmus were noted (Fig. 1c, d). Additionally, there was synpolydactyly with an additional hypoplastic toe between the fourth and fifth toes of the right foot (Fig. 1j). X-ray of the right foot demonstrated synpolydactyly with an extra hypoplastic metatarsal bone between the fourth and fifth metatarsals and an extra proximal phalangeal bone between the fourth and fifth toes (Fig. 1k). He had a complex congenital heart defect comprising double inlet left ventricle (DILV), L-malposition of great arteries, total anomalous pulmonary venous drainage (TAPVD), tubular hypoplasia of the aortic arch, and absence of interatrial septum that was surgically corrected after birth. Cranial ultrasound examination revealed hypoplasia of the corpus callosum. EEG showed signs of increased seizure susceptibility in the left temporo-occipital region. Furthermore, congenital bilateral cataract, sclerocornea, as well as hypospadias were noted. ABR demonstrated bilateral sensorineural hearing impairment. At age of 10 months, he presented with profound muscular hypotonia and severe global developmental delay. At the age of 19 months, he could not sit unaided, and had no speech development. At this age, his height and weight were within the normal range, but he was microcephalic (OFC: -4.3 SD). Clinical features of affected individuals 1–3 are summarized in Table 1. Extended clinical description of patients and pedigrees are provided as Supplemental data.
Truncating variants in MAPKAPK5 cause multiple congenital anomalies
Molecular analysis in family 1 comprised identification of chromosomal regions of homozygosity in affected individuals 1 and 2 by genome wide SNP array based hybridization along with ES of affected individual 1. Owing to the parental consanguinity, homozygous variants contained within blocks of homozygosity >1.5 Mb shared by the two sisters were prioritized. This revealed a homozygous 2-bp duplication, GeneBank NM_003668.3:c.207_208dupTG, in exon 4 of MAPKAPK5 (chr12[GRCh37]:g.112305396_112305397dup), predictably leading to a frameshift and generation of a stop codon seven triplets downstream; NP_003659.2:p.(Ala70Valfs*7) (Fig. 2a). This variant is not reported in gnomAD (v2.1.1).16 Sanger sequencing confirmed the c.207_208dupTG change in the homozygous state in individual 1 and in her affected sister (individual 2), while both unaffected parents carried this variant in the heterozygous state (Fig. S1a). The MAPKAPK5 variant was contained within a homozygous block of 20.38 Mb (chr12[GRCh37]:99285292-119670445) in affected individual 1 and 23.74 Mb (chr12:95931697-119674690) in affected individual 2.

(a) Schematic representation of the MAPKAPK5 gene structure illustrating location of the identified variants in affected individuals. MK5 functional domains and MK5 docking sites for interacting proteins are also indicated.31 NES nuclear export signal. NLS nuclear localization signal. (b) Relative quantification of MAPKAPK5 messenger RNA (mRNA) levels in fibroblasts from controls (C1, C2) and affected individual 1 (P1) by quantitative reverse transcription polymerase chain reaction (qRT-PCR). Results from two different MAPKAPK5 specific Taqman probes MAPKAPK5-572_m1 (Hs00177572_m1) and MAPKAPK5-300_m1 (Hs00934300_m1) are shown, n = 3. (c) Representative anti-MK5 and anti-ERK3 immunoblots in normal controls (C1 and C2) and affected individual 1 (P1) dermal fibroblasts (n = 3) showing absence of MK5 isoforms in affected individual 1 (duplet 52-50 kDa and 66 kDa). Asterisk on the blot designates a nonspecific band. Tubulin (TUB) served as protein loading control. The graph on the right shows relative quantification of ERK3 levels calculated by densitometry. For each blot, ERK3 densitometric values were normalized to tubulin (n = 3). In (b) and (c) graphs show means ± SD. ***P < 0.001; **P < 0.01; *P < 0.05. One-way analysis of variance (ANOVA) with Tukey´s multiple comparison test.
Trio ES in individual 3 also identified a homozygous 1-bp duplication NM_003668.3:c.1077dupT in exon 11 of MAPKAPK5 (chr12[GRCh37]:g.112326399dupT), predicted to result in a frameshift and premature stop of translation NP_003659.2:p.(Leu360Serfs*21) (Fig. 2a). This variant is also absent from the gnomAD control database (v2.1.1). Sanger sequencing validated the variant in the proband and both parents were confirmed to be heterozygotes (Fig. S1b). The two variants, p.(Ala70Valfs*7) and p.(Leu360Serfs*21), are predicted to lead to nonsense-mediated RNA decay (NMD).17
Loss of MK5 isoforms results in reduced levels of ERK3 and impaired F-actin formation
We developed dermal fibroblasts from affected individual 1 and examined the expression of MAPKAPK5 both at the messenger RNA (mRNA) and the protein level. MAPKAPK5 has been previously reported to be ubiquitously expressed.3,6 qRT-PCR analysis using two different Taqman probes showed approximately a 40% reduction of MAPKAPK5 transcript levels in patient fibroblasts compared with control cells, indicating that a proportion of the mutant transcript is likely degraded by NMD (Fig. 2b). We then analyzed the expression of MAPKAPK5 protein products (MK5). Anti-MK5 immunoblotting in control cells detected a double protein band of about 52–50 kDa corresponding to the expected molecular weight of MK5 (54 kDa, NM_003668.3; NM_139078.2) and a larger isoform of 66 kDa, all of which were absent in fibroblasts from the affected individual (Fig. 2c). The nature of the 66 kDa is unknown but a similar pattern of MK5 isoforms has been described in mice.18 As the MK5-interacting protein ERK3 is stabilized by MK5,9 we checked ERK3 protein expression in the same cultures. We found the amount of ERK3 to be diminished in fibroblasts from affected individual 1 in comparison with control cells, thus indicating that the c.207_208dupTG variant not only results in loss of function of MK5, but also significantly decreases the protein levels of ERK3 (Fig. 2c).
MK5 has also been reported to be involved in the polymerization of filamentous actin (F-actin).19 On this basis, we evaluated this process in primary fibroblasts from affected individual 1 and three different lines of normal control primary fibroblasts. We treated cells with latrunculin B to induce actin depolymerization for 30 minutes and after removal of this compound, we assessed the recovery of F-actin cytoskeleton at different time points by microscopy. Although there was some degree of variability in F-actin recovery between the different control cultures, in all experimental replicates the regeneration of F-actin was less efficient in the fibroblasts from affected individual 1 than in the control cells (Fig. 3). These data were further corroborated by confocal cytofluorescence quantification of F-actin and G-actin using phalloidin and DNase I double labeling (Figs. 4 and S2).

Representative microscopy images of primary fibroblasts from three different normal control cultures (C1, C2, and C3) and affected individual 1 (P1) untreated or treated with latrunculin B (2.5 μM, 30 minutes) and left to recover for different times. F-actin filaments were labeled with phalloidin-546 and nuclei were stained with DAPI. Scale bar = 25 μm. n = 6 (C2, P1), n = 4 (C1, C3). In all experiments, P1 cells showed worse and slower recovery of the actin cytoskeleton.

(a) Representative confocal images (Z-project) of control and patient fibroblasts (P1) costained with fluorescence phalloidin and DNase I for visualization of F-actin and G-actin, respectively. Fibroblasts were treated with latrunculin B (LatB) or its vehicle (DMSO) and fixed or left to recover for 30 minutes (latB + 30´). Untreated fibroblasts simultaneously seeded and cultured were also analyzed and used as controls. Green staining: phalloidin, red: DNase I, blue: DAPI. n = 4 except for C3 (n = 3). Scale bar = 50 μm. (b) F-actin/G-actin ratio in fibroblasts determined by fluorescence intensity. Normalized integrated density ratios calculated in cells from images such as those indicated in panel a are in the y-axis. For each line of fibroblasts, LatB values are referred to the mean value of the same cells treated with DMSO (left), and LatB+30´ values are referred to the mean value of the same cells in the untreated condition (right). Data are expressed as means ± SD. (c) Same as (b), but for F-actin integrated density (y-axis). F-actin and F-actin/G-actin integrated density values prior to normalization are shown in Supplementary material (Fig. S2). For each condition, n ≥ 44 (C3) and n ≥ 87 (C1, C2, P1) individual cells were analyzed in (b) and (c). Only statistical comparison between controls and P1 fibroblasts is shown. ***P < 0.001; **P < 0.01; *P < 0.05. Two-tailed Mann–Whitney test.
DISCUSSION
Our study identifies homozygous loss-of-function variants in MAPKAPK5 as the cause of a novel syndrome involving malformations of the heart, limbs, and severe neurodevelopmental disturbances. The three affected individuals with biallelic pathogenic variants in MAPKAPK5 had a congenital heart defect: two had a complex congenital heart defect (affected individuals 1 and 3) and the third had ASD and PDA. Psychomotor development was severely delayed in all three affected individuals, with no ability to walk. Two affected individuals did not achieve verbal communication, and the third individual used one single word only at age 9 years (affected individual 1). All three affected individuals shared EEG abnormalities but without seizures until the most recent examination. Brain anomalies were also present in all affected individuals; they were variable and included a dilated fourth ventricle, small cerebellar hemispheres, and hypoplasia of the corpus callosum. Variable eye anomalies, including optic disc pallor and tessellated retina, nystagmus, cataracts, and sclerocornea, were also seen in all individuals. Two individuals (affected individuals 1 and 3) were found with hearing impairment. Variable degrees of postaxial synpolydactyly of hands and feet were present in two individuals (affected individuals 1 and 3). The supernumerary, hypoplastic digit was consistently located between the fourth and fifth digits and may have branched from one of the middle or distal phalanges of the fifth ray or from metatarsal V. The supernumerary digit was always connected by cutaneous syndactyly to the fifth digit. Synpolydactyly type 1 (MIM 186000), type 2 (MIM 608180), and type 3 (MIM 610234) differ from the here described synpolydactyly because in the former the cutaneous syndactyly is consistently located between the middle and ring fingers with an additional finger in the syndactylous web in the hands. The type of synpolydactyly described here can be regarded as very specific and therefore probably helpful in clinical diagnosis of this syndrome. In addition, all affected individuals had in common facial dysmorphisms including a prominent forehead, narrow palpebral fissures, thin lips, and retrognathia. Two affected individuals were born with normal measurements, whereas one had a reduced birth weight (affected individual 1). Further, growth development was abnormal in two, who had short stature and failure to thrive (affected individuals 1 and 2), and two affected individuals developed postnatal microcephaly (affected individuals 2 and 3).
The physiological function of MK5 has been previously studied using two different Mapkapk5 mouse models generated by replacing exon 6 (∆ex6) or a major part of exon 8 (∆ex8) with the corresponding targeting cassettes.20,21 Homozygous ∆ex6 or ∆ex8 mice were found viable and fertile with no apparent abnormalities. However, ∆ex6 homozygous mutants exhibited embryonic lethality with incomplete penetrance (50%) on the C57/B6 background. As ∆ex6 homozygous mutants were underrepresented at least after E12 in intercrosses between C57/B6 ∆ex6 mice, developmental defects were suggested as the cause of these embryonic losses.9 Mapkapk5−/− mice (∆ex6) have also been characterized having impaired formation of dendritic spines in hippocampal neurons indicating a role of MK5 in neuronal morphogenesis and plasticity,22 which is consistent with the cognitive defects observed in the affected individuals reported here. In general, except for the aforementioned background-dependent embryonic lethality of ∆ex6 mice, the phenotypic consequences of pathogenic variants in MAPKAPK5 in human seem more severe than in mice, suggesting a more critical role of MK5 in the development of human neural tissue and heart that warrants the need for future studies.
It is worth mentioning that mouse embryonic fibroblasts derived from both ∆ex6 and ∆ex8 homozygotes were found to still be able to synthetize a residual proportion of mutant MK5 protein containing in-frame amino acid deletions that result from abnormal Mapkapk5 transcripts with skipping of exon 6 or exon 8, respectively.18 In contrast to this, western blot analysis of dermal fibroblasts from affected individual 1 using a C-terminal anti-MK5 antibody demonstrated absence of MK5 protein isoforms. However, we cannot exclude that the patient cells can synthetize a certain amount of N-terminal truncated MK5 protein (p.[Ala70Valfs*7]) since NMD was partially efficient in these cells. Patient fibroblasts were additionally characterized with a considerable reduction in ERK3 protein levels, a finding that was also observed in Mapkapk5−/− mice.9
Since impaired F-actin recovery after latrunculin B treatment was detected in patient dermal fibroblasts, our data support the previously recognized role of MK5 in regulating F-actin polymerization.19 Actin remodeling is a process necessary for key aspects of the cell, such as cell motility, cell morphology, or intracellular vesicle trafficking23 and is also essential for dendritic spine plasticity.24 In the adult mouse heart the MK5 protein is expressed in ventricular fibroblasts but not in ventricular myocytes,25 and Mapkapk5−/− ventricular fibroblasts have also been characterized with altered actin cytoskeleton and reduced motility.26 Thus, abnormal regulation of actin cytoskeleton dynamics due to loss of MK5 activity is likely contributing to the broad spectrum of clinical features observed in the patients of this report.
Further studies carried out in the ∆ex8 mouse model suggested that MK5 may have a tumor suppressor role by mediating the cellular senescence response to RAS signaling.21 Moreover, cell culture experiments indicated that MK5 acts downstream p38 to suppress RAS-MAPK signaling possibly by interfering with RAS-mediated activation of JNK.27,28 In this regard, there is only a limited degree of clinical overlap between the features of the affected individuals of this report and the phenotypic spectrum of RASopathies that are associated with pathogenic variants leading to hyperactivation of the RAS-MAPK signaling.29 RASopathy and MK5 affected individuals both have congenital heart defects and developmental delay. However, individuals with different forms of RASopathies show typical dysmorphic features that are different from those of the affected individuals of this report and do not share some other manifestations identified in affected individuals 1–3 such as brain anomalies, synpolydactyly, and microcephaly.
In conclusion, this work describes a severe phenotype associated with biallelic truncating pathogenic variants in MAPKAPK5 and reveals a central role of this gene in human development. Molecular and phenotypic characterization of more individuals with this entity will help to determine the phenotypic spectrum of pathogenic variants in MAPKAPK5.
Data availability
Data supporting this paper are contained within the article and Supplementary information. Any additional data not compromised by ethical issues will be available upon request.
References
Plotnikov, A., Zehorai, E., Procaccia, S. & Seger, R. The MAPK cascades: signaling components, nuclear roles and mechanisms of nuclear translocation. Biochim. Biophys. Acta 1813, 1619–1633 (2011).
Cargnello, M. & Roux, P. P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 75, 50–83 (2011).
Ni, H., Wang, X. S., Diener, K. & Yao, Z. MAPKAPK5, a novel mitogen-activated protein kinase (MAPK)-activated protein kinase, is a substrate of the extracellular-regulated kinase (ERK) and p38 kinase. Biochem. Biophys. Res. Commun. 243, 492–496 (1998).
Seternes, O. M. et al. Both binding and activation of p38 mitogen-activated protein kinase (MAPK) play essential roles in regulation of the nucleocytoplasmic distribution of MAPK-activated protein kinase 5 by cellular stress. Mol. Cell. Biol. 22, 6931–6945 (2002).
New, L., Jiang, Y. & Han, J. Regulation of PRAK subcellular location by p38 MAP kinases. Mol. Biol. Cell. 14, 2603–2616 (2003).
New, L. et al. PRAK, a novel protein kinase regulated by the p38 MAP kinase. EMBO J. 17, 3372–3384 (1998).
Aberg, E. et al. Regulation of MAPK-activated protein kinase 5 activity and subcellular localization by the atypical MAPK ERK4/MAPK4. J. Biol. Chem. 281, 35499–35510 (2006).
Kant, S. et al. Characterization of the atypical MAPK ERK4 and its activation of the MAPK-activated protein kinase MK5. J. Biol. Chem. 281, 35511–35519 (2006).
Schumacher, S. et al. Scaffolding by ERK3 regulates MK5 in development. EMBO J. 23, 4770–4779 (2004).
Seternes, O. M. et al. Activation of MK5/PRAK by the atypical MAP kinase ERK3 defines a novel signal transduction pathway. EMBO J. 23, 4780–4791 (2004).
Deleris, P. et al. Activation loop phosphorylation of ERK3/ERK4 by group I p21-activated kinases (PAKs) defines a novel PAK-ERK3/4-MAPK-activated protein kinase 5 signaling pathway. J. Biol. Chem. 286, 6470–6478 (2011).
Estan, M. C. et al. Recessive mutations in muscle-specific isoforms of FXR1 cause congenital multi-minicore myopathy. Nat. Commun. 10, 797 (2019).
Holtgrewe, M., et al. VarFish: comprehensive DNA variant analysis for diagnostics and research. Nucl. Acids Res. https://doi.org/10.1093/nar/gkaa241 (2020).
Ehmke, N. et al. Biallelic variants in KYNU cause a multisystemic syndrome with hand hyperphalangism. Bone. 133, 115219 (2020).
Kosmas, K. et al. CAP2 is a regulator of the actin cytoskeleton and its absence changes infiltration of inflammatory cells and contraction of wounds. Eur. J. Cell. Biol. 94, 32–45 (2015).
Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 581, 434–443 (2020).
Nagy, E. & Maquat, L. E. A rule for termination-codon position within intron-containing genes: when nonsense affects RNA abundance. Trends Biochem. Sci. 23, 198–199 (1998).
Ronkina, N. et al. Comparative analysis of two gene-targeting approaches challenges the tumor-suppressive role of the protein kinase MK5/PRAK. PLoS One 10, e0136138 (2015).
Tak, H. et al. 14-3-3epsilon inhibits MK5-mediated cell migration by disrupting F-actin polymerization. Cell. Signal. 19, 2379–2387 (2007).
Shi, Y. et al. Elimination of protein kinase MK5/PRAK activity by targeted homologous recombination. Mol. Cell. Biol. 23, 7732–7741 (2003).
Sun, P. et al. PRAK is essential for ras-induced senescence and tumor suppression. Cell. 128, 295–308 (2007).
Brand, F. et al. The extracellular signal-regulated kinase 3 (mitogen-activated protein kinase 6 [MAPK6])-MAPK-activated protein kinase 5 signaling complex regulates septin function and dendrite morphology. Mol. Cell. Biol. 32, 2467–2478 (2012).
Svitkina, T. The actin cytoskeleton and actin-based motility. Cold Spring Harb. Perspect. Biol. 10, a018267 (2018).
Fischer, M., Kaech, S., Knutti, D. & Matus, A. Rapid actin-based plasticity in dendritic spines. Neuron. 20, 847–854 (1998).
Nawaito, S. A. et al. MK5 haplodeficiency attenuates hypertrophy and preserves diastolic function during remodeling induced by chronic pressure overload in the mouse heart. Am. J. Physiol. Heart Circ. Physiol. 313, H46–H58 (2017).
Nawaito, S. A. et al. MK5 haplodeficiency decreases collagen deposition and scar size during post-myocardial infarction wound repair. Am. J. Physiol. Heart Circ. Physiol. 316, H1281–H1296 (2019).
Chen, G., Hitomi, M., Han, J. & Stacey, D. W. The p38 pathway provides negative feedback for Ras proliferative signaling. J. Biol. Chem. 275, 38973–38980 (2000).
Yoshizuka, N. et al. PRAK suppresses oncogenic ras-induced hematopoietic cancer development by antagonizing the JNK pathway. Mol. Cancer Res. 10, 810–820 (2012).
Tajan, M., Paccoud, R., Branka, S., Edouard, T. & Yart, A. The RASopathy family: consequences of germline activation of the RAS/MAPK pathway. Endocr. Rev. 39, 676–700 (2018).
Sobreira, N., Schiettecatte, F., Valle, D. & Hamosh, A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum. Mutat. 36, 928–930 (2015).
Moens, U. & Kostenko, S. Structure and function of MK5/PRAK: the loner among the mitogen-activated protein kinase-activated protein kinases. Biol. Chem. 394, 1115–1132 (2013).
Acknowledgements
We are grateful to patients and their parents for their participation in this study. The work at IIB was financially supported by the Spanish Ministry of Science, Innovation and Universities (PID2019-105620RB-I00/AEI/10.13039/501100011033 and SAF2016‐75434‐R (AEI/FEDER, UE). The authors also acknowledge the GeneMatcher tool30 which allowed the identification of these two families and Manuel Holtgrewe, Core Unit Bioinformatics of the Berlin Institute of Health (BIH), for processing of the exome data of family 2.
Author information
Authors and Affiliations
Contributions
Conceptualization: D.H., F.B., S.T., G.A.O., M.A., P.L., V.L.R.-P. Funding acquisition: V.L.R.-P. Formal analysis: E.F.-N., A.R.-B., N.E. Investigation: E.F.-N., R.G.-C., A.R.-B., J.N., V.L.R.-P., N.E., F.B. Project administration: P.L., V.L.R.-P. Resources: D.H., F.B., M.A., S.T., G.A.O., S.S. Supervision: D.H., F.B., S.T., M.A., V.L.R.-P. Visualization: E.F.-N., A.R.-B., G.A.O., M.A., D.H., F.B., V.L.R.-P. Writing—original draft: D.H., F.B., M.A., S.T., G.A.O., P.L., V.L.R.-P. Writing—review & editing: D.H., S.S., N.E., F.B., E.F.-N., A.R.-B., P.L., G.A.O., S.T., M.A., V.L.R.-P.
Corresponding authors
Ethics declarations
Ethics Declaration
Parental consent was obtained for all clinical and molecular studies of this report and for the publication of clinical photographs. All studies and investigations were performed according to the declaration of Helsinki principles of medical research involving human subjects and the study was approved by institutional Ethics Committees of National Research Centre, Hospital La Paz-CSIC and Charité-Universitaetsmedizin (EA2/140/17).
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Horn, D., Fernández-Núñez, E., Gomez-Carmona, R. et al. Biallelic truncating variants in MAPKAPK5 cause a new developmental disorder involving neurological, cardiac, and facial anomalies combined with synpolydactyly. Genet Med 23, 679–688 (2021). https://doi.org/10.1038/s41436-020-01052-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-020-01052-2
