
Abstract
Purpose:
A subset of patients with KIT/PDGFRA wild-type gastrointestinal stromal tumors show loss of function of succinate dehydrogenase, mostly due to germ-line mutations of succinate dehydrogenase subunits, with a predominance of succinate dehydrogenase subunit A. The clinical outcome of these patients seems favorable, as reported in small series in which patients were individually described. This work evaluates a retrospective survival analysis of a series of patients with metastatic KIT/PDGFRA wild-type succinate dehydrogenase–deficient gastrointestinal stromal tumors.
Methods:
Sixty-nine patients with metastatic gastrointestinal stromal tumors were included in the study (11 KIT/PDGFRA wild-type, of whom 6 were succinate dehydrogenase deficient, 5 were non–succinate dehydrogenase deficient, and 58 were KIT/PDGFRA mutant). All six succinate dehydrogenase–deficient patients harbored SDHA mutations. Kaplan–Meier curves and log-rank tests were used to compare the survival of patients with succinate dehydrogenase subunit A–mutant gastrointestinal stromal tumors with that of KIT/PDGFRA wild-type patients without succinate dehydrogenase deficiency and patients with KIT/PDGFRA-mutant gastrointestinal stromal tumors.
Results:
Follow-up ranged from 8.5 to 200.7 months. The difference between succinate dehydrogenase subunit A–mutant gastrointestinal stromal tumors and KIT/PDGFRA-mutant or KIT/PDGFRA wild-type non–succinate dehydrogenase deficient gastrointestinal stromal tumors was significant considering different analyses (P = 0.007 and P = 0.033, respectively, from diagnosis of gastrointestinal stromal tumor for the whole study population; P = 0.005 and P = 0.018, respectively, from diagnosis of metastatic disease for the whole study population; P = 0.007 for only patients who were metastatic at diagnosis).
Conclusion:
Patients with metastatic KIT/PDGFRA wild-type succinate dehydrogenase–deficient gastrointestinal stromal tumors harboring succinate dehydrogenase subunit A mutations present an impressively long survival. These patients should be identified in clinical practice to better tailor treatments and follow-up over time.
Genet Med 17 5, 391–395.
Similar content being viewed by others
Main
A subset of patients with KIT/PDGFRA wild-type gastrointestinal stromal tumors (GISTs) show loss of function of succinate dehydrogenase (SDH), resulting in negative immunohistochemical staining for the SDH subunit B (SDHB) protein.1,2,3,4,5,6 In ~40 to 50% of cases, the cause of the SDH deficiency is germ-line mutation in any of subunits A, B, C, or D, although mutations in SDHA are predominant.7 In the remaining cases, the molecular cause of tumorigenesis and SDH deficiency is unknown. Other molecular events correlated with SDH complex deficiency in GIST have recently been described. Epigenomic studies suggest a correlation between succinate metabolism and tumor genomic methylation.8 Moreover, a microRNA profiling study showed that wild-type SDHB-immunonegative tumors present a distinct pattern compared with KIT/PDGFRA-mutant tumors.9 The common clinical and pathological features of patients with SDH-deficient GIST have been widely described.10 According to a small series in which patients were individually described, these patients seem to have favorable clinical outcomes even after the development of metastases.6,7 To date, however, no survival analysis of these patients has been explored. The aim of this work was to evaluate a survival analysis of adult patients with metastatic KIT/PDGFRA wild-type SDH-deficient GIST as compared with patients with KIT/PDGFRA wild-type non–SDH-deficient and KIT/PDGFRA-mutant GIST to better define the trend of their clinical outcomes in the metastatic setting.
Materials and Methods
Patients and tumors
Sixty-nine patients with metastatic GIST who came to our clinic from 2004 were retrospectively evaluated and included in the study (mean age, 56.2 years (range: 27–79 years); 34 female, 35 male; 31 stomach, 33 small-intestine, and 5 other tumors).
Nonmetastatic patients and patients with localized GIST surgically removed without recurrence were excluded because their prognosis is highly affected by other factors, including mitotic count and the anatomic site rather than the SDH or KIT/PDGFRA genotype. Eleven patients presented with KIT/PDGFRA wild-type GIST and 58 with KIT/PDGFRA-mutant GIST. In all patients with KIT/PDGFRA wild-type GIST, screening for other tumors, paraganglioma, or pheochromocytoma was performed during the follow-up for GIST, and family and personal history were assessed. Patients with GIST and a known genetic syndrome were excluded because their disease commonly has a different natural course than that of sporadic cases.10
For the patients with KIT/PDGFRA wild-type GIST, SDH-deficient status was assessed by immunohistochemical negativity for the SDHB protein and by SDH genome sequencing of all four subunits. Six of 11 patients had SDH-deficient GIST, and 5 patients had non–SDH-deficient GIST. All six patients with SDH-deficient GIST had SDHA-mutant GIST, and all cases except two have been previously described.2,4,7 None of the five patients with KIT/PDGFRA wild-type non–SDH-deficient GIST had mutations in any SDH complex subunits.
Forty-nine of 58 patients with KIT/PDGFRA-mutant GIST (84%) had a GIST with primary mutations in KIT exon 11, and 9 patients (16%) had a GIST with a primary mutation in KIT exon 9. Exon 18 PDGFRA D842V–mutant GISTs were excluded because they are resistant to tyrosine kinase inhibitors, which may introduce a bias in a survival study of metastatic GIST.
Statistical analysis
The survival analysis focused on the patients with metastatic SDH-deficient GIST (in this study, all patients harbored SDHA mutations hereafter referred to as SDHA mutant GIST), as compared with patients with metastatic KIT/PDGFRA wild-type GIST without SDH deficiency and patients with metastatic KIT/PDGFRA-mutant GIST. Kaplan–Meier curves using log-rank tests were developed for the overall survival (OS) of all three groups of patients. The survival analyses examined (i) the time from diagnosis of GIST to the death of the patient or the last follow-up for the whole study population, (ii) the time from diagnosis of metastatic disease (at first diagnosis or at recurrence) to the death of the patient or the last follow-up for the whole study population, and (iii) for cases with metastases at diagnosis, the time from diagnosis of GIST to the death of the patient or the last follow-up.
Results
The characteristics and clinical outcomes of patients with KIT/PDGFRA wild-type GIST (both SDHA-mutant GIST and non–SDH-deficient GIST) are listed in Table 1 (overall mean age: 39.7 years (range: 19–65 years); mean age of patients with SDHA-mutant GIST: 27 years (range: 19–39 years); and mean age of patients with non–SDH-deficient GIST: 55 years (range: 41–65 years)). All patients with SDHA-mutant GIST except one presented with metastatic disease at the time of diagnosis.
Therapeutic management included the following: Two patients (GIST_07 and GIST_145) received three lines of therapy (imatinib, sunitinib, and nilotinib (now ongoing)), and both underwent primary debulking surgery during sunitinib treatment; three patients underwent surgery of primary tumors and metastases at the time of diagnosis and, of them, one patient (GIST_10) then received only imatinib (now ongoing); one patient (GIST_150) received imatinib and then sunitinib (now ongoing); and one patient (ID_9) did not receive any tyrosine kinase inhibitor therapy. Only one patient with SDHA-mutant GIST (GIST_151) presented with localized disease at diagnosis, underwent radical surgery of the primary tumor, and at recurrence received imatinib (now ongoing). All patients with KIT/PDGFRA wild-type GIST without SDH deficiency presented with localized disease at the time of diagnosis and underwent surgery of primary tumors. At recurrence, the therapeutic management of these patients included only tyrosine kinase inhibitors, and only one patient (GIST_127) underwent surgery for a solitary hepatic lesion at first recurrence.
Twenty-seven of 58 patients with KIT/PDGFRA-mutant GIST (46%) had a localized GIST at diagnosis and then presented a recurrence, whereas 31 of 58 (54%) presented with metastatic disease at diagnosis. The treatment of these patients was commonly according to standard guidelines. All of them received at least first-line therapy with imatinib, 32 patients (55%) received second-line therapy with sunitinib, and 13 patients (22%) received a third-line therapy (nilotinib, regorafenib, or sorafenib).
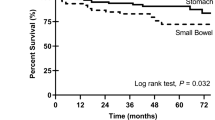
Follow-up data were available for all patients (OS ranged between 8.5 and 200.7 months). During this period no deaths were observed in the group with SDHA mutations, 3 of 5 deaths occurred in the group with KIT/PDGFRA wild-type non–SDH-deficient GIST, and 28 of 58 deaths occurred in the group with KIT/PDGFRA-mutant GIST. The OS curves from diagnosis of GIST are reported in Figure 1a ; the difference between the group with SDHA-mutant GIST and the groups with KIT/PDGFRA-mutant or KIT/PDGFRA wild-type non–SDH-deficient GIST was significant (P = 0.007 and 0.033, respectively, log-rank test). The OS curves considered from the diagnosis of metastatic disease are reported in Figure 1b ; the difference between the group with SDHA-mutant GIST and the groups with KIT/PDGFRA-mutant GIST or KIT/PDGFRA wild-type non–SDH-deficient GIST was significant (P = 0.005 and 0.018, respectively, log-rank test). The OS curves for only the patients who had metastatic disease at diagnosis, thus excluding the KIT/PDGFRA wild-type non–SDH-deficient group—in which all patients had localized disease at diagnosis—are reported in Figure 1c ; the difference between the group with SDHA-mutant GIST and the group with KIT/PDGFRA-mutant GIST was significant (P = 0.007, log-rank test).

Overall survival curves of patients with metastatic succinate dehydrogenase (SDH)–deficient gastrointestinal stromal tumor (GIST) (in this study all patients harbored SDH subunit A (SDHA) mutations) as compared with patients with metastatic KIT/PDGFRA wild-type GIST without SDH deficiency and patients with KIT/PDGFRA-mutant GIST. (a) Time from diagnosis of GIST to the death of the patient or the last follow-up for the whole study population. (b) Time from diagnosis of metastatic disease (at first diagnosis or at recurrence) to the death of the patient or the last follow-up for the whole study population. (c) Time from diagnosis of GIST to the death of the patient or the last follow-up only for cases with metastases at diagnosis.
Discussion
According to a small series in which patients were individually described, patients with SDH-deficient GIST seem to have a favorable clinical outcome. Because the aim of this study was to describe the clinical course of patients with advanced SDH-deficient GIST, we evaluated OS in the metastatic setting. We did not analyze progression-free survival or the response rate for the following reasons: (i) the aim of this study was not to correlate sensitivity to single tyrosine kinase inhibitors with SDH status, which would require a large number of patients, even if some published data on sunitinib activity are reported for this subset of patients11,12; (ii) most of the patients with SDH-deficient GIST received different lines of therapies (standard and/or experimental), and the sample size for each treatment was too small for any meaningful progression-free survival analysis or response rate evaluation; and (iii) two of our patients experienced impressive long-term disease stabilization for years during third-line treatment with nilotinib, which we suppose likely represents the natural history of the disease rather than an effect of the therapy administered.13
The survival analysis showed that SDHA mutations were associated with a better clinical outcome as compared with KIT/PDGFRA mutations and KIT/PDGFRA wild-type without SDH deficiency. The difference in survival between the group with SDHA mutations and the group with KIT/PDGFRA mutations was significant in all survival analyses (from diagnosis of primary tumors and from diagnosis of metastatic disease). These findings are extremely interesting if we consider that these patients had metastatic disease at diagnosis and they did not have a theoretical chance of cure due to their resistance to imatinib, which is the most efficacious targeted treatment in patients with GIST.
Also, the difference in survival between the group with SDHA mutations and the group with KIT/PDGFRA wild-type without SDH deficiency was significant. Moreover, we emphasize that the difference in OS values and clinical outcomes between the two groups should be considered substantial. In the follow-up period, all patients with SDHA mutations are still alive and have an extremely long OS, ranging from 74 to 200 months (~6 to 16.5 years), except for patient ID_9, in whom GIST was diagnosed only 24 months ago ( Table 1 ). OS of patients without SDH deficiency ranged from 35 to 128 months ( Table 1 ). Among these patients, however, survival of patients with longer follow-up was influenced by additional favorable clinical features, such as a long disease-free period between the removal of primary tumors and recurrence, and the use of surgery for a single hepatic lesion at first recurrence in the patient with 128 months of follow-up (GIST_127). Unfortunately, our survival findings are limited to KIT/PGDFRA wild-type SDH-deficient GIST harboring SDHA mutations and do not extend to the whole SDH-deficient GIST family. Large numbers of patients are needed to show any survival differences between the several SDH-deficient groups, such as those with mutations in other SDH subunits or those in which the cause of SDH deficiency does not depend on SDH gene mutations.
The indolent course of disease for metastatic patients with SDHA mutations suggests the importance of recognizing these patients in clinical practice. First, all patients carried a germ-line first-hit mutation in SDHA that required a second-hit somatic mutation to develop the tumor. So, given that mutations of the SDH genes are germ-line in patients without a personal or family history of paraganglioma or other tumors, these patients may be carriers of an attenuated form of Carney triad and Carney–Stratakis syndrome or a novel, as yet unknown syndrome. Therefore, considering the long survival in patients with metastatic disease, it is reasonable that all patients with KIT/PDGFRA wild-type SDHA mutant GIST should be genetically tested and strictly monitored over time for the development of other tumors.
Second, these patients with metastatic disease could benefit from alternative therapeutic approaches that do not adhere at all to standard guidelines, for example, primary surgical debulking or any interval surgery, for which, as is well known, the benefit is higher in patients with stable or responding disease as compared with patients with focal progressing disease. However, the indolent nature of the tumors in patients with SDHA mutations may make them suitable for such strategies also if they develop a mild progression.14,15
Third, the loss of SDHA and, in general, the SDH complex may induce a pseudo-hypoxic status, leading to the activation of several nuclear genes involved in angiogenesis and proliferation through similar molecular pathways, as has been observed in renal cell cancers that display loss of von Hippel–Lindau tumor suppressor function.16 Although the aim of the current study was not the evaluation of progression-free survival or the response rate to single treatments, for which larger series are necessary, all three patients with SDHA mutations with unresectable metastatic disease (GIST_07, GIST_10, and GIST_150) ( Table 1 ) demonstrated resistance to imatinib and had prolonged disease control on sunitinib. Therefore, SDHA-mutant GIST could be amenable to anti-angiogenetic inhibitors for as long as possible and probably also in patients with disease progression and toxicity that may require optimizations of the schedule or dosage.
Finally, a correlation between the overexpression of the insulin-like growth factor receptor 1 protein and the status of SDH complex deficiency has been reported.17,18 Despite the small number of patients with this molecular background reported, the data are mature enough to examine whether insulin-like growth factor receptor 1 could be considered a target for trials with insulin-like growth factor receptor 1 inhibitors in these selected patients.
In conclusion, our findings provide further evidence that patients with KIT/PDGFRA wild-type SDH-deficient GIST harboring SDHA mutations experience good survival outcomes and confirm the necessity of identifying these patients in practice using a simple immunohistochemistry test for SDHB and genetic testing for all patients who are SDHB negative because their clinical management in terms of treatments and follow-up may benefit from a more patient-tailored approach.
Disclosure
M.A.P. on behalf of GIST Study Group Bologna: Novartis Oncology supported GIST research programs.
References
Janeway KA, Kim SY, Lodish M, et al.; NIH Pediatric and Wild-Type GIST Clinic. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc Natl Acad Sci USA 2011;108:314–318.
Pantaleo MA, Astolfi A, Indio V, et al. SDHA loss-of-function mutations in KIT-PDGFRA wild-type gastrointestinal stromal tumors identified by massively parallel sequencing. J Natl Cancer Inst 2011;103:983–987.
Italiano A, Chen CL, Sung YS, et al. SDHA loss of function mutations in a subset of young adult wild-type gastrointestinal stromal tumors. BMC Cancer 2012;12:408.
Dwight T, Benn DE, Clarkson A, et al. Loss of SDHA expression identifies SDHA mutations in succinate dehydrogenase-deficient gastrointestinal stromal tumors. Am J Surg Pathol 2013;37:226–233.
Gill AJ, Chou A, Vilain RE, Clifton-Bligh RJ . “Pediatric-type” gastrointestinal stromal tumors are SDHB negative (“type 2”) GISTs. Am J Surg Pathol 2011;35:1245–1247; author reply 1247.
Miettinen M, Killian JK, Wang ZF, et al. Immunohistochemical loss of succinate dehydrogenase subunit A (SDHA) in gastrointestinal stromal tumors (GISTs) signals SDHA germline mutation. Am J Surg Pathol 2013;37:234–240.
Pantaleo MA, Astolfi A, Urbini M, et al.; GIST Study Group. Analysis of all subunits, SDHA, SDHB, SDHC, SDHD, of the succinate dehydrogenase complex in KIT/PDGFRA wild-type GIST. Eur J Hum Genet 2014;22:32–39.
Killian JK, Kim SY, Miettinen M, et al. Succinate dehydrogenase mutation underlies global epigenomic divergence in gastrointestinal stromal tumor. Cancer Discov 2013;3:648–657.
Kelly L, Bryan K, Kim SY, et al.; NIH Pediatric and Wild-Type GIST Clinic. Post-transcriptional dysregulation by miRNAs is implicated in the pathogenesis of gastrointestinal stromal tumor [GIST]. PLoS One 2013;8:e64102.
Pasini B, McWhinney SR, Bei T, et al. Clinical and molecular genetics of patients with the Carney-Stratakis syndrome and germline mutations of the genes coding for the succinate dehydrogenase subunits SDHB, SDHC, and SDHD. Eur J Hum Genet 2008;16:79–88.
Rege TA, Wagner AJ, Corless CL, Heinrich MC, Hornick JL . “Pediatric-type” gastrointestinal stromal tumors in adults: distinctive histology predicts genotype and clinical behavior. Am J Surg Pathol 2011;35:495–504.
Janeway KA, Albritton KH, Van Den Abbeele AD, et al. Sunitinib treatment in pediatric patients with advanced GIST following failure of imatinib. Pediatr Blood Cancer 2009;52:767–771.
Pantaleo MA, Nannini M, Saponara M, et al.; GIST Study Group, University of Bologna, Bologna, Italy. Impressive long-term disease stabilization by nilotinib in two pretreated patients with KIT/PDGFRA wild-type metastatic gastrointestinal stromal tumours. Anticancer Drugs 2012;23:567–572.
Raut CP, Posner M, Desai J, et al. Surgical management of advanced gastrointestinal stromal tumors after treatment with targeted systemic therapy using kinase inhibitors. J Clin Oncol 2006;24:2325–2331.
Gronchi A, Fiore M, Miselli F, et al. Surgery of residual disease following molecular-targeted therapy with imatinib mesylate in advanced/metastatic GIST. Ann Surg 2007;245:341–346.
Linehan WM, Srinivasan R, Schmidt LS . The genetic basis of kidney cancer: a metabolic disease. Nat Rev Urol 2010;7:277–285.
Nannini M, Astolfi A, Paterini P, et al. Expression of IGF-1 receptor in KIT/PDGF receptor-α wild-type gastrointestinal stromal tumors with succinate dehydrogenase complex dysfunction. Future Oncol 2013;9:121–126.
Belinsky MG, Rink L, Flieder DB, et al. Overexpression of insulin-like growth factor 1 receptor and frequent mutational inactivation of SDHA in wild-type SDHB-negative gastrointestinal stromal tumors. Genes Chromosomes Cancer 2013;52:214–224.
Acknowledgements
Special thanks to the GIST Study Group members, University of Bologna, Bologna, Italy: Annalisa Altimari, Federica Bertolini, Paolo Castellucci, Massimo Del Gaudio, Monica Di Battista, Stefano Fanti, Walter Franco Grigioni, Elisa Gruppioni, Paola Paterini, Paola Tomassetti, and Maurizio Zompatori.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Pantaleo, M., Lolli, C., Nannini, M. et al. Good survival outcome of metastatic SDH-deficient gastrointestinal stromal tumors harboring SDHA mutations. Genet Med 17, 391–395 (2015). https://doi.org/10.1038/gim.2014.115
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2014.115
Keywords
This article is cited by
-
REGISTRI: Regorafenib in first-line of KIT/PDGFRA wild type metastatic GIST: a collaborative Spanish (GEIS), Italian (ISG) and French Sarcoma Group (FSG) phase II trial
Molecular Cancer (2023)
-
Succinate dehydrogenase deficient gastrointestinal stromal tumor in a three month old boy with a fatal clinical course: a case report and review of literature
Diagnostic Pathology (2021)
-
Preferential MGMT methylation could predispose a subset of KIT/PDGFRA-WT GISTs, including SDH-deficient ones, to respond to alkylating agents
Clinical Epigenetics (2019)
-
The progressive fragmentation of the KIT/PDGFRA wild-type (WT) gastrointestinal stromal tumors (GIST)
Journal of Translational Medicine (2017)
-
Gene expression identifies heterogeneity of metastatic behavior among gastrointestinal stromal tumors
Journal of Translational Medicine (2016)
