
Abstract
Purpose: Although Lynch syndrome is characterized by marked genetic heterogeneity, some specific mutations are observed at high frequency in well-defined populations or ethnic groups due to founder effects.
Methods: Genomic breakpoint identification, haplotype analysis, and mutation age determination were performed in 14 unrelated patients and 95 family members presenting the same MLH1 exonic rearrangement, among a series of 84 Lynch syndrome families with germline mutations in MLH1, MSH2, or MSH6.
Results: All 14 probands harbored an identical deletion, comprising exons 17–19 of the MLH1 gene and exons 26–29 of the LRRFIP2 gene, corresponding to the MLH1 mutation c.1896 + 280_oLRRFIP2:c.1750-678del. This mutation represents 17% of all deleterious mismatch repair mutations in our series. Haplotype analysis showed a conserved region of approximately 1 Mb, and the mutation age was estimated to be 283 ± 78 years. All 14 families are originated from the Porto district countryside.
Conclusion: We have identified a novel MLH1 exonic rearrangement that is a common founder mutation in Lynch syndrome families, indicating that screening for this rearrangement as a first step may be cost-effective during genetic testing of Lynch syndrome suspects of Portuguese ancestry, especially those originating from the Porto district.
Similar content being viewed by others


Main
Lynch syndrome is a highly penetrant, autosomal dominant disease characterized by early-onset colorectal cancer (CRC) and extracolonic tumors of the endometrium, stomach, small bowel, ureter, renal pelvis, ovary, and hepatobiliary tract.1 Families are usually selected for genetic testing using the Amsterdam criteria or the Bethesda guidelines.2,3 Although the former are rather specific and allow selection of families for direct germline mutation analysis, the latter have higher sensitivity but lower specificity and require a prescreening by microsatellite instability analysis or immunohistochemistry for mismatch repair (MMR) proteins in tumor tissue.4,5
The genetic defect underlying Lynch syndrome is a germline mutation in one of the four MMR genes MLH1, MSH2, MSH6, and PMS2.6 Approximately 85% of the mutations described are found in MSH2 and MLH1, with MSH6 and PMS2 mutations accounting for the remaining 10% and 5%, respectively (International Collaborative Group on HNPCC Mutation Database, http://www.insight-group.org). The mutational spectrum of Lynch syndrome includes mainly point mutations, small insertions, and deletions, as well as changes affecting splice sites. However, the use of new techniques allowed the discovery that a significant proportion of pathogenic alterations are large genomic rearrangements, in most cases single or multiexonic deletions or duplications inactivating MLH1 or MSH2.7 Based on the October 2009 Human Gene Mutation database, MLH1 and MSH2 exonic deletions/duplications represented 21% of all reported mutations.8
Although Lynch syndrome can be originated by many different mutations located throughout the four relevant MMR genes, specific mutations are observed at high frequency in well-defined populations or ethnic groups due to founder effects. For example, founder mutations have been identified in Lynch syndrome families from China, the United States, Italy, and among Ashkenazi Jews.9–12 The identification of founder mutations facilitates the molecular diagnosis of Lynch syndrome by making cost-effective mutational analysis to specific gene regions before full screening of all MMR genes. We herein present a novel MLH1 exonic rearrangement that is a founder mutation in Portuguese Lynch syndrome families.
MATERIALS AND METHODS
Patients, samples, and DNA extraction
This study includes 14 Lynch syndrome families presenting the same MLH1 exonic rearrangement, from a total series of 84 families with pathogenic MLH1, MSH2, or MSH6 germline mutations (data not shown), all of which have been identified by routine genetic diagnosis during the period of 1997 to 2009 at the Genetics Department of the Portuguese Oncology Institute, Porto, Portugal, after genetic counseling and informed consent. Seven families were followed up at the Portuguese Oncology Institute, six at the S. João Hospital, and one at the Padre Américo Hospital, all located in the Porto district. Nine of the 14 families fulfilled the Amsterdam criteria, whereas the remaining presented the Bethesda criteria for genetic testing. After written informed consent, DNA was isolated from peripheral blood samples from the 14 index individuals and subsequently from 95 family members, using the salt–chloroform extraction method.13 The geographic origin of these families was inferred from the birthplace of the oldest carrier or of the oldest affected family member most likely to be a carrier.
Microsatellite instability and MMR immunohistochemical analyses
In all nine families that fulfilled the Amsterdam criteria and in three of the five cases presenting the Bethesda criteria (because tumor sample was not available), MMR mutation screening was performed directly from the blood sample of the index case. In the remaining two families with Bethesda criteria and available tumor sample, microsatellite instability and MMR immunohistochemical analyses were performed in the carcinoma sample of one index case and on a tubulovillous adenoma from the second index case. Additionally, MLH1 immunoexpression was assessed in four additional carcinomas from three families with Amsterdam criteria (one index case from one family, one index case and one affected relative from a second family, and one affected relative from a third family).
Microsatellite instability evaluation was performed using the Bethesda panel of markers (BAT25, BAT26, D2S123, D5S346, and D17S250) and the 1997 National Cancer Institute guidelines. Polymerase chain reaction (PCR) was carried out as previously described using fluorescence-labeled primers.14 Fragments were analyzed for length variations on an ABI Prism 310 DNA sequencer (Applied Biosystems, Foster City, CA), and allele sizes were determined using Genemapper software (version 3.7, Applied Biosystems). The results were independently scored by two observers, and a second round of analyses confirmed the results.
Assessment of MLH1, MSH2, MSH6, and PMS2 immunoexpression was performed as described previously.15
Screening for MLH1 and MSH2 germline alterations
The 14 index individuals had initially been screened for mutations in MLH1 and MSH2 coding exons (except the acceptor splice site of MLH1 exon 12, MSH2 exon 1, and the acceptor splice site of MSH2 exon 5) by denaturing gradient gel electrophoresis (DGGE), using primers and conditions as described by Ingeny (The Netherlands) and Wu et al.16 Fragments with abnormal DGGE patterns and the acceptor splice site of MLH1 exon 12, MSH2 exon 1, and the acceptor splice site of MSH2 exon 5 were analyzed by direct sequencing in an ABI PRISM 310 automatic sequencer using Big Dye Terminator Chemistry (Applied Biosystems), according to the manufacturer's recommendations. The 14 index cases and the 95 family members reported in this study were then screened for MSH2 and MLH1 exonic deletions and duplications by multiplex ligation-dependent probe amplification (MLPA), according to the instructions of the SALSA MLPA P003 and P248 MLH1/MSH2 kits (MRC-Holland, Amsterdam).
Genomic breakpoint identification
The strategy for breakpoint identification was based on the heterozygosity status information obtained from a set of microsatellite (including the ones used in the haplotype studies, see later) and single-nucleotide polymorphism (SNP) markers (Fig., Supplemental Digital Content 1, http://links.lww.com/GIM/A182). Subsequently, primers were designed spanning the putative breakpoints, and long-range PCR was carried out using the Expand Long Template PCR System (Roche Diagnostics, Mannheim, Germany), using conditions recommended by the manufacturer. PCR fragments containing the suspected weight were sequenced with BigDye Terminator cycle sequencing chemistry on an ABI PRISM 310 automatic sequencer (Applied Biosystems), according to the manufacturer's recommendations.
The deletion nomenclature is in agreement with the rules recommended by the Human Genome Variation Society (www.hgvs.org/mutnomen). Genomic breakpoint locations are given using the reference sequences NM_000249 and NM_006309 for MLH1 and LRRFIP2, respectively.
Design of mutation-specific assay
After breakpoint identification, we designed a single-nucleotide primer extension assay to detect this MLH1 rearrangement. First, a three-primer PCR selective amplification was developed in which the mutated allele is amplified with primers MLH1-INT16F 5′-AAATTGATGAGGTGTGACAGCCATTCT-3′ (forward) and LRRFIP2-INT25R 5′-AAGGACAGCTGGGGAAGCCA-3′ (reverse) and the normal allele with the same forward primer and the reverse primer MLH1-INT16R 5′-GGCCTGCAGGGATTCGGCTC-3′. PCR reactions were performed in a 20 μL reaction containing 30–50 ng of DNA, 2 μL of 10x Taq reaction buffer, 1.5 μL of MgCl2 (1.875 mM), 1 μL of deoxynucleoside triphosphate mix (250 μM deoxythymidine triphosphate, 250 μM dexyadenosine triphosphate, 250 μM deoxyguanosine triphosphate, and 250 μM deoxycytidine triphosphate, Applied Biosystems), 0.2 pmol/μL of primer MLH1-INT16F, 0.1 pmol/μL of primers LRRFIP2-INT25R and MLH1-INT16R, and 0.75 units of Taq DNA polymerase (Fermentas). After a 95°C preincubation step for 10 minutes, PCR was performed in a total of 35 cycles using the following conditions: 95°C denaturation for 30 seconds, annealing at 58°C for 45 seconds, and extension at 72°C during 45 seconds, followed by 10 minutes of final extension at 72°C.
After PCR, we performed a multiplexed nucleotide primer extension reaction using dye label terminators (SNaPshot kit, Applied Biosystems). The primers were designed in the forward direction, with one annealing immediately 5′ to the first nucleotide of the breakpoint region questioning both the wild-type and the mutated alleles (BKP1F 5′-GAGGTAGAAGTTGCAGTGA-3′) and the second (BKP2-WTF 5′-GACTGACGTAGAAGTTGCAGTGAGC-3′) and the third primers (BKP2-MTF 5′-GACTGACGTAGAAGTTGCAGTGACC-3′) being specific to the wild-type and mutated alleles, respectively, and questioning the third nucleotide of the breakpoint region (Fig., Supplemental Digital Content 2, http://links.lww.com/GIM/A183). The multiplex primers were designed to be of different lengths using a nonhomologous PolydGACT tail at the 5′ end, so that they could be distinguished by size during capillary electrophoresis separation. The SNaPshot reaction was performed with 3, 2, and 1 pmol/μL of primers BKP1F, BKP2-WTF, and BKP2-MTF, respectively.
Analysis of breakpoints sequence context
Breakpoints were defined as a set of coordinates on the genome spanning the genomic sequence of the deletion. Bioinformatics analyses were carried out to analyze the genomic context of the region. Using the RepeatMasker software, low-complexity DNA sequences and interspersed repeats were searched in both MLH1 intron 16 and LRRFIP2 intron 25.
Microsatellite and SNP typing
A total of 14 probands and 95 family members were genotyped for polymorphic microsatellite markers flanking MLH1, namely D3S1609, D3S1612, D3S1561, TR89812, D3S1611, TR100328, D3S1298, D3S3527, and D3S3522. The order of the markers, the consensus repeat, and the distances relative to each other and to MLH1 are shown in Fig., Supplemental Digital Content 1, http://links.lww.com/GIM/A182. The physical distances of the genetic markers were derived from National Center for Biotechnology Information Map Viewer (http://www.ncbi.nlm.nih.gov/projects/mapview/). The consensus pattern was obtained with the software Tandem Repeats Finder (http://www.tandem.bu.edu/). The primer sequences for the amplification of the markers were derived from the Human Genome database (http://www.gdb.org), except for two new markers (TR89812 and TR100328) that were designed with the Primer express software. All nine markers were assayed by PCR using fluorescently end-labeled primers. PCR products were run on an ABI PRISM 310 Genetic Analyzer together with the fluorescence labeled DNA fragment size standard TAMRA (Applied Biosystems). Genotyping of two intragenic SNP located within MLH1 exon 8 (c.655A>G) and intron 14 (c.1668-19A>G) was performed by DGGE.
SNP markers were used to obtain a haplotype spanning approximately 2.6 Mb encompassing the region between TR89812 and D3S3527 microsatellite markers, where the first recombinant and/or mutational events were observed. To capture most of the genetic variation in this region and to avoid redundant SNP markers (i.e., markers in strong linkage disequilibrium), we performed Tag-SNP, namely Tagger Multimarker, using International HapMap Project CEPH (Utah residents with ancestry from northern and western Europe) population data (www.hapmap.org). We developed SNaPshot assays for 19 SNP markers by multiplexed nucleotide primer extension reaction using dye label terminators (Applied Biosystems). The primers for multiplex amplification and single base extension (Table, Supplemental Digital Content 3, http://links.lww.com/GIM/A184) were designed using the online Primer-BLAST tool (http://www.ncbi.nlm.nih.gov/tools/primer-blast/). AutoDimer (www.cstl.nist.gov/strbase/NIJ/AutoDimer.htm) was used to test for potential hairpin structures and primer dimer problems. The 19 SNPs were PCR amplified in four multiplex reactions with amplicon lengths between 101 bp and 381 bp. Amplification was carried out in a 9700 Thermocycler (Applied Biosystems). After a 95°C preincubation step for 5 minutes, PCR was performed in a total of 35 cycles using the following conditions: 95°C denaturation for 30 seconds, annealing at 55°C for 30 seconds, and extension at 72°C during 30 seconds, followed by 10 minutes of final extension at 72°C. The multiplex SNaPshot reaction and capillary electrophoresis were done following the manufacturer's protocol (Applied Biosystems).
Haplotype construction and estimation of mutation age
Haplotype construction was performed manually based on the genotypes obtained from index cases and family members. We estimated the age of the mutation from the variation accumulated in their ancestral haplotypes, as described by Martins et al.17 This method takes into account both recombination (c) and mutation (μ) rates in the generation of variation. The probability of change per generation (ε) is given by ε = 1 − ([1 − c] [1 − μ]), and the average of mutation and recombination events (λ) equals εt, where t is the number of generations. The recombination rate (c) was estimated from the physical distance between the two most distant markers (D3S1609 and D3S3522) using a conversion factor calculated in Rutgers Map Interpolator (http://compgen.rutgers.edu/old/map-interpolator/). The estimate of average mutation rate used was 7.8 × 10−4 for dinucleotides markers.18
RESULTS
Identification of a novel MLH1 exonic rearrangement
Analysis of the constitutional blood-derived DNA by MLPA in the 14 index cases reported herein revealed a reduction of the peak signal for exons 17, 18, and 19 of MLH1 of approximately 50% compared with normal controls, suggesting a heterozygous genomic deletion of these exons (Fig. 1A). Subsequent analysis with a MLPA confirmation kit revealed that exon 26 of the LRRFIP2 gene downstream of MLH1 was also deleted in all 14 cases (Fig. 1A). This MLH1 c.1897-?_2271+?del (HGVS, NM_000249: initiating codon = 1) mutation is present in approximately 17% (14/84) of all Lynch syndrome families with pathogenic mutations identified at the Genetics Department of Portuguese Oncology Institute of Porto (unpublished data).

Molecular characterization of the MLH1 c.1896 + 280_oLRRFIP2:c.1750-678del mutation. A, Capillary electrophoresis pattern from one case (blue) presenting a signal reduction of approximately 50% for exons 17, 18, and 19 of MLH1 gene (arrows) compared with a normal control (green) detected by MLPA using the SALSA MLPA P003 kit (upper panel) and the kit P248 MLH1/MSH2 (lower panel), the latter showing also a signal reduction for LRRFIP2 exon 26 (arrows). B, Long-range PCR with primers spanning the putative breakpoints revealed a 741 bp fragment (arrow) in the cases with the MLH1 mutation (Lane 1). Lane 2 shows a negative case. NTC is a nontemplate control and MW refers to 100 bp DNA standard. C, Sequence electrophorogram of the 741 bp PCR fragment showing the breakpoint region of the mutated allele. D, Scheme representing the MLH1 c.1896 + 280_oLRRFIP2:c.1750-678del mutation found in all index cases, with the breakpoint downstream of MLH1 exon 16 and upstream of LRRFIP2 exon 25.
Genomic breakpoint identification
The MLH1 c.1897-?_2271+?del (HGVS, NM_000249: initiating codon = 1) mutation was fully characterized on the nucleotide level. After long-range PCR with primers spanning the putative breakpoints, a 741 bp fragment appeared in the cases with the MLH1 mutation (Fig. 1B). Sequence analysis of this PCR product revealed the breakpoint region in the mutated allele (Fig. 1C). All 14 probands harbored an identical 11,627 bp deletion, comprising exons 17, 18, and 19 of the MLH1 gene and exons 26, 27, 28, and 29 of the adjacent LRRFIP2 gene (Fig. 1D). The 5′ and 3′ breakpoints were located 280 bp downstream of MLH1 exon 16 and 678 upstream of LRRFIP2 exon 25, respectively. Therefore, the full description of the MLH1 mutation is c.1896 + 280_oLRRFIP2:c.1750-678del (HGVS, NM_000249: initiating codon = 1).
Mutation-specific detection
Genomic DNA amplification by the three-primer set in the cases presenting the MLH1 deletion resulted in two fragments of 533 bp and 551 bp from the wild-type and mutated alleles, respectively, whereas in the negative cases only the 533 bp fragment from the wild-type allele appears (Fig., Supplemental Digital Content 2, http://links.lww.com/GIM/A183). After multiplexed nucleotide primer extension reaction, the positive cases present the wild-type (G) and the mutant (C) nucleotides with the BKP1F primer and the wild-type (C) and the mutant (A) nucleotides with the BKP2-WTF and BKP2-MTF primers, respectively (Fig., Supplemental Digital Content 2, http://links.lww.com/GIM/A183). The negative cases only present the wild-type G and C nucleotides (Fig., Supplemental Digital Content 2, http://links.lww.com/GIM/A183). The SNaPshot reaction was performed on all 14 index cases and in 20 negative cases previously analyzed by direct sequencing, and all the cases were concordant.
Breakpoints sequence analysis
The genomic sequences flanking the deletion breakpoints in MLH1 intron 16 and LRRFIP2 intron 25 were analyzed for low-complexity DNA sequences and interspersed repeats, and one AluSx repeat and one AluSc repeat, respectively, were found at the breakpoints.
MMR immunohistochemical and microsatellite instability analyses
All five carcinomas and the tubulovillous adenoma studied by immunohistochemistry showed absence of MLH1 expression (Fig. 2). Additionally, both the carcinoma and the tubulovillous adenoma of the two index cases with Bethesda criteria showed high microsatellite instability (all five markers presented instability).

Representative image of MLH1 immunostain: stromal cells show distinct nuclear immunoreactivity, whereas adenocarcinoma cells (right side) are negative.
Ancestral STR-based haplotypes and age estimate
Eight different haplotypes were phased for 10 of the 14 families. The results of the haplotype analyses for the 10 informative families are shown in Figure 3, and the most parsimonious relationships among flanking haplotypes are presented as a phylogenetic network in Figure 4. The probability of mutation versus recombination was evaluated considering the number of stepwise mutations required and intermediate haplotypes observed. In the 10 informative families, SNP haplotypes were constructed to establish whether a specific microsatellite was different from the consensus because of a recombination event rather than a mutation (Fig., Supplemental Digital Content 4, http://links.lww.com/GIM/A185).

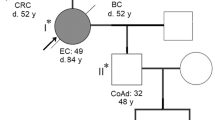
Simplified pedigrees of the 10 informative families and haplotype results (A to J). The order of the microsatellite and SNP markers is D3S1609, D3S1612, D3S1561, TR89812, c.655A>G, D3S1611, c.1668-19A>G, TR100328, D3S1298, D3S3527, and D3S3522 (from top to bottom), and the alleles that segregate with the mutation are underlined. Unaffected individuals are indicated with open symbols, patients affected with colorectal cancer with black symbols, and breast cancer is represented by striped circles. The oblique line indicates that the patient is deceased. Plus and minus signals represent family members with and without the MLH1 c.1896 + 280_oLRRFIP2:c.1750-678del mutation, respectively.

Phylogenetic network showing the most parsimonious relationships among flanking short tandem repeat-based haplotypes in families carrying the MLH1 c.1896 + 280_oLRRFIP2:c.1750-678del mutation. Circle and line sizes are proportional to the number of families and stepwise mutations, respectively, and diamonds indicate recombination events.
Haplotype analysis of the 10 informative families revealed a conserved region of approximately 1 Mb. Based on the mutation and recombination events observed in microsatellite haplotypes and assuming a generation time of 25 years, the age estimate for the MLH1 mutation c.1896 + 280_oLRRFIP2:c.1750-678del (HGVS, NM_000249: initiating codon = 1) is 283 ± 78 years (Table 1).
The geographic origins of the MLH1 mutation positive families are shown in Figure 5, all being originated from the district of Porto, Portugal, but away from the most densely populated areas (the city of Porto or surrounding cities). The remaining 70 Lynch families identified at our institution have a much disperse geographic origin from the entire North and Center of Portugal.

Geographic origin of the families with the MLH1 c.1896 + 280_oLRRFIP2:c.1750-678del germline mutation in Portugal. Black circles and the number within represent the families and its frequency. All families are originated from a small region of the district of Porto (shadowed region at the right) but not from the city of Porto itself, which is located as indicated by the white circle.
Clinicopathologic associations
CRC was the most frequent malignancy (73%) observed in the families presenting the MLH1 exonic rearrangement reported herein, followed by stomach (7%) and endometrial cancer (5%). The median age of diagnosis of CRC was 44 years. Seven patients presented metachronous and one patient presented synchronous colorectal carcinomas. The histomorphological study of all five colorectal carcinomas that could be evaluated were adenocarcinomas (one poorly and four moderately to well differentiated), and mucinous production was observed in three of the carcinomas. Three of the five carcinomas were located in the right colon.
DISCUSSION
The novel MLH1 mutation c.1896 + 280_oLRRFIP2:c.1750-678del that we report herein was identified in 14 Portuguese Lynch syndrome families, representing approximately 17% (14/84) of all deleterious MMR mutations and approximately 41% (14/34) of the MLH1 mutations detected at the Genetics Department of Portuguese Oncology Institute of Porto (unpublished data). All the families presenting this mutation have their origin in a small geographic area in the north of Portugal, comprising several counties in the periphery of the Porto district. At least 20% (17/84) of all Lynch syndrome families with a pathogenic mutation identified at our institution have their origin from other regions of Portugal, and none of the 14 families with this MLH1 rearrangement originated from the most densely populated areas of Porto district. Furthermore, none of the previous publications on Portuguese Lynch families mostly from South Portugal reported this exonic rearrangement.19,20 These data indicate that our finding of this mutation in 14 families from the Porto district is not explained by referral bias. In other countries, the deletion of exons 17–19 of the MLH1 gene has been reported in one Lynch syndrome family from Taiwan21 and another from France.22 However, none of these studies described the genomic breakpoints or deletion of LRRFIP2 exon 26 (detectable by the MLPA P248 MLH1/MSH2 kit), so one can assume that the genomic rearrangement reported herein is novel. In fact, it has recently been shown that the breakpoints of the MLH1 rearrangement reported in the Lynch syndrome family from Taiwan21 are different from the ones we describe in this study (Dr. Ling-Ling Hsieh, Chang Gung University, Taiwan, personal communication, 2010).
Haplotype analysis by microsatellite and SNP markers flanking the MLH1 gene in the 10 informative families revealed a conserved region of approximately 1 Mb, indicating that these families indeed share a common ancestor. Based on the mutation and recombination events observed in microsatellite haplotypes and assuming a generation time of 25 years, the origin of this mutation could be traced back to the beginning of the 18th century. This relatively young age is in agreement with the confined geographic origin of the 14 families bearing the MLH1 c.1896 + 280_oLRRFIP2:c.1750-678del mutation. Furthermore, the same exact breakpoint was identified in each of the families, which is also a strong indicator of a common origin. The available data, therefore, indicates that this is a novel exonic rearrangement involving deletion of the last three exons of MLH1 and the last four exons of the contiguous LRRFIP2 gene, being a frequent founder mutation in Lynch families originated from the Porto district in North Portugal.
Several studies have shown that genomic deletions and duplications in the MSH2 or MLH1 genes are a frequent cause of Lynch syndrome.7,9,23–27 Wijnen et al.25 reported a frequency of 6.5% of MSH2 deletions in Lynch families from a Dutch population. More recently, large MMR gene rearrangements have been reported in 11–15% of Lynch syndrome families in France,26 The Netherlands,7 and Germany,27 in all instances most commonly in MSH2. The higher frequency of large genomic rearrangements in MSH2 is presumably due to a higher Alu density (34.2% on average).26 Alu repeats are short interspersed elements whose transposition has been repeatedly implicated in genetic variability and heritable disorders, including Lynch syndrome and hereditary breast/ovarian cancer.27,28 In the MLH1 gene, Alu-mediated exonic deletions have previously been reported mainly associated with a founder effect, as the deletion of exon16 represents approximately 50% of all MLH1 mutations in the Finnish population.23 This Finnish founder mutation, a 3.5 kb MLH1 deletion, resulted from a recombination event between two Alu repeats located in introns 15 and 16.23 Mauillon et al.24 also observed a deletion of exons 13–16 of the MLH1 gene, caused by a recombination event between two Alu repeats located in introns 12 and 16. As we found Alu repeats around the breakpoints of both MLH1 intron 16 and LRRFIP2 intron 25, Alu-mediated homologous recombination might also have been involved in the origin of the Portuguese founder exonic rearrangement that we report in this study.
The MLH1 protein forms a heterodimer with MLH3, PMS2, or PMS1 and recruits other DNA repair proteins to the MMR complex for the excision and repair of DNA.29 The MLH1 exons deleted in the rearrangement that we describe (exons 17–19) code for the MLH3, PMS2, or PMS1-binding domain. The carcinoma and the adenoma analyzed for microsatellite status presented high instability, which is indicative of MMR deficiency. Moreover, all the tumors evaluated for MMR immunoexpression lacked MLH1 protein expression, demonstrating that this large rearrangement leads to loss of protein. On the other hand, this rearrangement affects also the adjacent LRRFIP2 gene, causing deletion of the last four exons (26–29). LRRFIP2 was recently identified as a modulator of the Wingless-type mouse mammary tumor virus integration site family (Wnt) signaling pathway.30 LRRFIP2 interacts with disheveled (Dvl) to increase the cellular abundance of β-catenin and activates LEF/TCF-dependent gene transcription of Wnt target genes. It presents a coiled-coil domain at its carboxyl terminus and a serine-rich region at the amino terminus.31 Liu et al.30 analyzed the molecular function of LRRFIP2 with a series of deleted mutants and observed that the activity of LRRFIP2 was severely reduced after truncation of either the carboxyl terminus or the amino terminus, indicating that both domains are required for its function. These authors also demonstrated that a mutant form of LRRFIP2 containing only the amino terminus acts as a dominant negative form and abolishes the activities of both LRRFIP2 and Dvl.30 The LRRFIP2 exons deleted in the rearrangement we here present code for the LRRFIP2 coiled-coil domain at its carboxyl terminus. If this rearrangement results in a truncated protein that exerts a dominant negative effect on the wild-type LRRFIP2, this would result in decreased β-catenin levels, which is the opposite of what is found in most CRCs. Further studies are warranted to clarify the role of this LRRFIP2 germline mutation, if any, in the context of Lynch syndrome. On the other hand, our 14 Lynch syndrome families show typical features of this disease, such as predominance of right colon carcinomas, early onset, high microsatellite instability, and lack of MLH1 expression in tumor tissue, thereby indicating that the relevant genetic defect underlying Lynch syndrome in these families is the inactivation of MLH1 gene through this large exonic rearrangement.
Because of the high frequency of the MLH1 c.1896 + 280_oLRRFIP2:c.1750-678del mutation in our population, we developed a mutation-specific assay to screen for this mutation quickly and inexpensively, which involves a three-primer PCR-specific amplification assay followed by a single-nucleotide primer extension reaction. Although the MLPA technique allows identification of the deletion of MLH1 exons 17, 18, and 19 while screening for all MLH1 and MSH2 exonic rearrangements, it requires another methodology to fully characterize this specific mutation. Using our approach, the breakpoint region of the MLH1 and LRRFIP2 deletion is interrogated in a single, multiplex reaction, providing two independent assessments of the mutation in question. This mutation-specific assay is a faster and less expensive method for the detection of this rearrangement as it involves only a standard PCR followed by a SNaPshot reaction. In addition, this method is highly flexible, and more mutations can be added to the multiplex reaction.
In conclusion, we have identified a novel MLH1 exonic rearrangement that is a common founder mutation in Lynch syndrome families originated from the Porto district in North Portugal. This rearrangement corresponds to a large deletion involving MLH1 exons 17–19 and LRRFIP2 exons 26–29, which has presumably resulted from homologous recombination between two Alu sequences present in introns 16 and 25 of the genes MLH1 and LRRFIP2, respectively. The high proportion of the MLH1 c.1896 + 280_oLRRFIP2:c.1750-678del mutation indicates that screening for this rearrangement as a first step may be cost-effective during genetic testing of Lynch syndrome suspects of Portuguese ancestry, especially those originating from the Porto district.
REFERENCES
Lynch HT, de la Chapelle A . Hereditary colorectal cancer. N Engl J Med 2003; 348: 919–932.
Vasen HF, Watson P, Mecklin JP, Lynch HT . New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology 1999; 116: 1453–1456.
Umar A, Risinger JI, Hawk ET, Barrett JC . Testing guidelines for hereditary non polyposis colorectal cancer. Nat Rev Cancer 2004; 4: 153–158.
Baudhuin LM, Burgart LJ, Leontovich O, Thibodeau SN . Use of microsatellite instability and immunohistochemistry testing for the identification of individuals at risk for Lynch syndrome. Fam Cancer 2005; 4: 255–265.
Vasen HF, Möslein G, Alonso A, et al. Guidelines for the clinical management of Lynch syndrome (hereditary non-polyposis cancer). J Med Genet 2007; 44: 353–362.
Lagerstedt Robinson K, Liu T, et al. A. Lynch syndrome (hereditary nonpolyposis colorectal cancer) diagnostics. J Natl Cancer Inst 2007; 99: 291–299.
Gille JJ, Hogervorst FB, Pals G, et al. Genomic deletions of MSH2 and MLH1 in colorectal cancer families detected by a novel mutation detection approach. Br J Cancer 2002; 87: 892–897.
Stenson PD, Mort M, Ball EV, et al. The Human Gene Mutation Database: 2008 update. Genome Med 2009; 1: 13.
Chan TL, Yuen ST, Ho JW, et al. A novel germline 1.8-kb deletion of hMLH1 mimicking alternative splicing: a founder mutation in the Chinese population. Oncogene 2001; 20: 2976–2981.
Wagner A, Barrows A, Wijnen JT, et al. Molecular analysis of hereditary nonpolyposis colorectal cancer in the United States: high mutation detection rate among clinically selected families and characterization of an American founder genomic deletion of the MSH2 gene. Am J Hum Genet 2003; 72: 1088–1100.
Caluseriu O, Di Gregorio C, Lucci-Cordisco E, et al. A founder MLH1 mutation in families from the districts of Modena and Reggio-Emilia in northern Italy with hereditary non-polyposis colorectal cancer associated with protein elongation and instability. J Med Genet 2004; 41: 34.
Sun S, Greenwood CM, Thiffault I, Hamel N, Chong G, Foulkes WD . The HNPCC associated MSH2*1906G–>C founder mutation probably originated between 1440 CE and 1715 CE in the Ashkenazi Jewish population. J Med Genet 2005; 42: 766–768.
Müllenbach R, Lagoda PJ, Welter C . An efficient salt-chloroform extraction of DNA from blood and tissues. Trends Genet 1989; 5: 391.
Dietmaier W, Wallinger S, Bocker T, Kullmann F, Fishel R, Rüschoff J . Diagnostic microsatellite instability: definition and correlation with mismatch repair protein expression. Cancer Res 1997; 57: 4749–4756.
Pinheiro M, Ahlquist T, Danielsen SA, et al. Colorectal carcinomas with microsatellite instability display a different pattern of target gene mutations according to large bowel site of origin. BMC Cancer 2010; 10: 587.
Wu Y, Berends MJ, Mensink RG, et al. Association of hereditary nonpolyposis colorectal cancer-related tumors displaying low microsatellite instability with MSH6 germline mutations. Am J Hum Genet 1999; 65: 1291–1298.
Martins S, Calafell F, Gaspar C, et al. Asian origin for the worldwide-spread mutational event in Machado-Joseph disease. Arch Neurol 2007; 64: 1502–1508.
Gyapay G, Morissette J, Vignal A, et al. The 1993-94 Généthon human genetic linkage map. Nat Genet 1994; 7: 246–339.
Sousa R, Lage P, Ferreira S, et al. Need of new clinical criteria for the identification of genetic Lynch syndrome. Acta Med Port 2007; 20: 535–542.
Ferreira S, Lage P, Sousa R, et al. Familial colorectal cancer type X: clinical, pathological and molecular characterization. Acta Med Port 2009; 22: 207–214.
Tang R, Hsiung C, Wang JY, et al. Germ line MLH1 and MSH2 mutations in Taiwanese Lynch syndrome families: characterization of a founder genomic mutation in the MLH1 gene. Clin Genet 2009; 75: 334–345.
Rouleau E, Lefol C, Bourdon V, et al. Quantitative PCR high-resolution melting (qPCR-HRM) curve analysis, a new approach to simultaneously screen point mutations and large rearrangements: application to MLH1 germline mutations in Lynch syndrome. Hum Mutat 2009; 30: 867–875.
Nyström-Lahti M, Kristo P, Nicolaides NC, et al. Founding mutations and Alu-mediated recombination in hereditary colon cancer. Nat Med 1995; 1: 1203–1206.
Mauillon JL, Michel P, Limacher JM, et al. Identification of novel germline hMLH1 mutations including a 22 kb Alu-mediated deletion in patients with familial colorectal cancer. Cancer Res 1996; 56: 5728–5733.
Wijnen J, van der Klift H, Vasen H, et al. MSH2 genomic deletions are a frequent cause of HNPCC. Nat Genet 1998; 20: 326–328.
Charbonnier F, Olschwang S, Wang Q, et al. MSH2 in contrast to MLH1 and MSH6 is frequently inactivated by exonic and promoter rearrangements in hereditary nonpolyposis colorectal cancer. Cancer Res 2002; 62: 848–853.
Wang Y, Friedl W, Lamberti C, et al. E. Hereditary nonpolyposis colorectal cancer: frequent occurrence of large genomic deletions in MSH2 and MLH1 genes. Int J Cancer 2003; 103: 636–641.
Peixoto A, Santos C, Rocha P, et al. The c. 156_157insAlu BRCA2 rearrangement accounts for more than one-fourth of deleterious BRCA mutations in northern/central Portugal. Breast Cancer Res Treat 2009; 114: 31–38.
Mitchell RJ, Farrington SM, Dunlop MG, Campbell H . Mismatch repair genes hMLH1 and hMSH2 and colorectal cancer: a HuGE review. Am J Epidemiol 2002; 156: 885–902.
Liu J, Bang AG, Kintner C, et al. Identification of the Wnt signaling activator leucine-rich repeat in Flightless interaction protein 2 by a genome-wide functional analysis. Proc Natl Acad Sci USA 2005; 102: 1927–1932.
Fong KS, de Couet HG . Novel proteins interacting with the leucine-rich repeat domain of human flightless-I identified by the yeast two-hybrid system. Genomics 1999; 58: 146–157.
Acknowledgements
This study was supported by the Portuguese Health Ministry and Liga Portuguesa Contra o Cancro. The authors acknowledge Dr. Ling-Ling Hsieh for confirming that the breakpoints of the MLH1 deletion of exons 17–19 they reported in one Lynch syndrome family from Taiwan21 are different from the mutation they present in this study.
Author information
Authors and Affiliations
Corresponding author
Additional information
Disclosure: The authors declare no conflict of interest.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Web site (www.geneticsinmedicine.org).
Rights and permissions
About this article
Cite this article
Pinheiro, M., Pinto, C., Peixoto, A. et al. A novel exonic rearrangement affecting MLH1 and the contiguous LRRFIP2 is a founder mutation in Portuguese Lynch syndrome families. Genet Med 13, 895–902 (2011). https://doi.org/10.1097/GIM.0b013e31821dd525
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1097/GIM.0b013e31821dd525
Keywords
This article is cited by
-
Epithelioid fibrous histiocytoma: three diagnostically challenging cases with novel ALK gene fusions, unusual storiform growth pattern, and a prominent spindled morphology
Virchows Archiv (2022)
-
A survey of the clinicopathological and molecular characteristics of patients with suspected Lynch syndrome in Latin America
BMC Cancer (2017)
-
Co-occurrence of nonsense mutations in MSH6 and MSH2 in Lynch syndrome families evidencing that not all truncating mutations are equal
Journal of Human Genetics (2016)
-
The role of germline mutations in the BRCA1/2 and mismatch repair genes in men ascertained for early-onset and/or familial prostate cancer
Familial Cancer (2016)
-
Target gene mutational pattern in Lynch syndrome colorectal carcinomas according to tumour location and germline mutation
British Journal of Cancer (2015)
