
Abstract
Purpose
Inherited cataract, opacification of the lens, is the most common worldwide cause of blindness in children. We aimed to identify the genetic cause of isolated autosomal-dominant lamellar cataract in a five-generation British family.
Methods
Whole exome sequencing (WES) was performed on two affected individuals of the family and further validated by direct sequencing in family members.
Results
A novel missense mutation NM_001040667.2:c.190A>G;p.K64E was identified in the DNA-binding-domain of heat-shock transcription factor 4 (HSF4) and found to co-segregate with disease.
Conclusion
We have identified a novel mutation in HSF4 in a large British pedigree causing dominant congenital lamellar cataract. This is the second mutation in this gene found in the British population. This mutation is likely to be dominant negative and affect the DNA-binding affinity of HSF4.
Similar content being viewed by others

Introduction
Cataract, the cloudiness of the lens is the most frequent cause of blindness worldwide, representing almost half of all causes of blindness globally: the WHO estimates that 18 million people are bilaterally blind from cataract.1 Congenital cataracts are seen in 1–15 per 10 000 births in the United Kingdom and are a significant cause of childhood visual impairment.2, 3 Congenital cataract can occur in isolation or in association with other non-ocular manifestations and is a predominant feature in >200 genetic disorders. Congenital cataract may be familial and display considerable genotypic and phenotypic heterogeneity.4 Inheritance is most commonly autosomal dominant (AD), usually with complete penetrance but with highly variable expressivity. The phenotypic classification of the cataract depends on the position and type of the lens opacity such as: anterior polar, posterior polar, nuclear, lamellar, coralliform, blue-dot (cerulean), cortical, pulverulent, polymorphic, complete cataract, and posterior nuclear cataract.5, 6 Significant progress has been made in identifying the molecular genetic basis of human cataract. More than 40 genes have been recruited, including the large family of crystallins encoding transparent intracellular lens proteins, membrane gap junction proteins (connexins), water channel proteins (aquaporins), solute carrier protein, cytoskeletal proteins, transmembrane proteins, lens intrinsic membrane protein, chromatin-modifying protein-4B, receptor tyrosine kinase gene EPH receptor A2,7 an endoplasmic reticulum membrane-embedded protein, Wolframin,8 and transcription factor genes (eg, PAX6, FOXE3, EYA1, MAF, PITX3, and HSF4).7
Herein we report a novel mutation in the heat-shock transcription factor 4 (HSF4) gene causing an isolated autosomal-dominant lamellar cataract in a five-generation British family.
Methods
Phenotyping
In this study, the family was identified through the proband attending the Genetic Service at Moorfields Eye Hospital, London, UK. Local ethics committee approval was obtained and all of the participants gave written informed consent. All the family members underwent full ophthalmic examination, including slit lamp examination; all affected individuals were diagnosed as having isolated lamellar cataract.
Whole exome sequencing (WES) and bioinformatics analysis
Genomic DNA was extracted from EDTA sequestered blood samples taken with informed consent and local ethical approval using the Nucleon II DNA Extraction Kit (Scotlab Bioscience, Strathclyde, Scotland, UK). The DNA was sequenced at AROS Applied Biotechnology (Aarhus, Denmark). Exon capture and target enrichment was performed using the SureSelectXT Human All Exon V5 kit (Agilent, Santa Rosa, CA, USA). Paired-end sequencing was ran on an Illumina Hiseq 2500 high-throughput sequencer (Illumina, San Diego, CA, USA) generating mean exome coverage of 50 ×. Raw data in fastq format was analysed using the Phenopolis platform.9 The short read sequence data were aligned using novoalign (version 3.02.08). Variants and indels were called according to GATK best practices (joint variant calling followed by variant quality score recalibration).10 The variants were then annotated using the Variant Effect Predictor (VEP).11 Variants were then filtered to only contain novel variants not present in public control databases Kaviar12 and gnomAD (http://gnomad.broadinstitute.org/) and predicted to be moderately or highly damaging according to the VEP. Cosegregation of the filtered variants in both affected individuals was then performed. Finally, the list of variants was further screened using Phenopolis for genes associated with the Human Phenotype Ontology12, 13 term ‘lamellar cataract’ (HP:0007971) according to OMIM.14
Structural bioinformatics
The protein structure of HSF4 was analysed using SWISS-MODEL15 (https://swissmodel.expasy.org/repository/uniprot/Q9ULV5). The best PDB16 match, with a match of 75%, was the structure of the DNA-binding-domain (DBD) HSF1 (PDB ID 2LDU, solved with NMR by the Northeast Structural Genomics Consortium Target HR3023C 2011). An X-ray-solved structure of HSF1 DBD interacting with DNA was also found in PDB (PDB ID 5D5U17. All structures were downloaded in PDB format and analysed using Pymol (version 1.8) locally. The Pymol Mutagenesis tool was used to visualise the effect of the mutation on the HSF1–DNA complex.
Sanger sequencing
Bi-directional direct Sanger sequencing was performed to validate the variant identified by next-generation sequencing. Genomic DNA was amplified by PCR using GoTaq 2X master mix (AB gene; Thermo Scientific, Epsom, UK) and HSF4-specifc primers designed with Primer3 http://bioinfo.ut.ee/primer3-0.4.0/primer3/. PCR conditions were as follows: 94 °C for 10 min of initial denaturation followed by 30 cycles of amplification of 30 s at 94 °C, 30 s at 60 °C, and 45 s at 72 °C. After the PCR products were reacted with BigDye Terminator v3.1, they were run on ABI 3730 Genetic Analyzer (both from Applied Biosystems, Foster City, CA, USA) and analyzed using SeqMan Pro (version 8.0.2 from DNASTAR) sequence analysis. After validating the variant, family segregation was performed in all the individuals. Further, 96 unrelated patients from our autosomal-dominant congenital cataract (ADCC) panel were screened for this variant by bi-directional Sanger sequencing to look for any other occurrence of the mutation.
Results
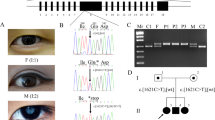
A large five-generation pedigree comprising 16 members, including 9 affected individuals, 3 unaffected individuals, and 4 spouses, were examined and all affected members had evidence of lamellar cataract (Figure 1).

Abridged pedigree of the British family with lamellar cataract. Squares and circles symbolize males and females, respectively. Open and filled symbols indicate unaffected and affected individuals, respectively.
WES was undertaken in two affected individuals (IV-1, IV-6). Variant annotation and filtering was performed using the Phenopolis platform. From a total of 149 133 variants in the family, 43 441 were found to co-segregate in the two affected individuals. A filter for novel variants that were moderately or highly damaging was then applied and returned 79 variants. After gene panel screening, a rare heterozygous damaging variant, NM_001040667.2:c.190A>G, in HSF4 on chromosome 16q22.1 was identified. Direct sequencing confirmed that the missense mutation c.190A>G in exon 4 of HSF4 co-segregated with all the affected members of the family (Figure 2).

Sequence analysis of HSF4. An unaffected individual (upper chromatogram illustrates a normal control and a missense mutation c.190A>G shown in few affected members of the family with lamellar cataract.
This single base change is predicted to result in a lysine (K) to glutamic acid (E) amino-acid substitution (p.K64E) in the highly conserved DBD, as shown on the protein structure of HSF4 (Figure 3a). The structural change on HSF4 induced by the mutation was estimated to be negligible according to the difference in the PDB models returned by SWISS-MODEL (root mean square deviation of 0.009). However, the homologous amino acid has a hydrogen bond to the DNA molecule in the structure of DNA-binding complex of the homologous protein HSF1 (Figure 3b). The mutation from a lysine to a glutamic acid is likely to affect that bond, as the lysine is positively charged, whereas glutamic acid is negatively charged, which could have some effect on the binding affinity to the DNA molecule (Figure 3c). Further in silico analysis and experimental work is required to verify this claim.

Structure of the DNA-binding domain of HSF4. (a) Known mutations and their sidechains are displayed in red. The K64E mutation is just downstream of an alpha helix and is a point of contact with the DNA molecule. (b and c) Point of contact of the amino acid (red) in the DBD interacting with the DNA molecule (orange and blue). (b) In the wild-type protein, the amino acid is lysine. (c) In the mutant protein, the amino acid is a glutamic acid.
Discussion
Here we report a missense mutation NM_001040667.2:c.190 A>G in the HSF4 gene on chromosome 16q22.1 in a five-generation English pedigree with congenital lamellar cataract. All the affected family members had bilateral congenital cataract and age at onset varied from birth to age 5–7 years. One of the patients IV-6 also had glaucoma.
Lamellar cataract is also referred to as zonular cataract and is one of the commonest phenotypes of ADCC. The inner fetal nucleus is made up of a clear lens surrounded by an opacified shell that is, in turn, surrounded by clear cortex, which may contain opacities referred to as ‘riders’ or ‘cortical spokes’. Lamellar cataract represents a disturbance in the lens development at a particular time and the cataractous ‘shell’ varies in size according to the stage of fetal development at which the disturbance occurs.5 HSF4 is expressed in both cell types (epithelial cells and fibre cells) at an early stage of lens development and is required for normal cell growth and differentiation of these two cell types.18
HSF4 belongs to the family of heat-shock transcription factors that regulate the expression of heat-shock proteins in response to various stresses, such as high temperature, infection, and free radicals, and also in development. HSF4 expression is tissue specific and has two splice forms, (i) HSF4a, which inhibits transcription of other heat-shock factor genes by binding directly to the heat-shock element and (ii) HSF4b, which contains an additional 30 amino acids and acts as an activator of transcription. HSF4 comprises 15 exons and encodes a heat-shock transcriptional protein of 493 amino-acid residues with a DBD, an amino-terminal hydrophobic repeat (HR/A-B), an isoform-specific region and downstream of hydrophobic repeat (DHR).19, 20
As described in Table 1, 16 different mutations in HSF4 have been identified in different ethnicities for both for autosomal-dominant and -recessive congenital cataract, as well as age-related cataract.21, 22 Eight of these have been found in the Chinese population, three autosomal-recessive mutations in Pakistani families,23 and one splice site mutation in a large Tunisian family.24 Recently, Benham et al25 reported a novel homozygous mutation in an Iranian family causing congenital cataract. So far, only two mutations have been found in Europeans; in Danish families, p.R119C was a recurrent mutation; and a novel mutation p.H35Y in a British family with no description of the cataract phenotype.26 Each of these mutations were found in autosomal-dominant cataract, including our novel heterozygous mutation p.K64E in a large British pedigree with lamellar cataract.
Interestingly, all known autosomal-dominant mutations in HSF4 lie within the α-helical DBD, whereas the recessive mutations lie outside this highly conserved functional domain.27
These observations support that mutations in the DBD are dominant negative rather than loss of function. The binding affinity of HSF4 to the DNA might be perturbed that could interfere with the wild-type molecule. This could potentially disrupt the transcription factor ‘orchestra’ in eye development, leading to abnormal lens cells conducive to protein aggregates and hence the onset of cataract. It has also been confirmed that HSF4 regulates lens fibre cell differentiation by modulating the expression of certain lens structural proteins, such as lens-specific crystallins, beaded filament proteins, and fibroblast growth factors.18 Further studies have shown that HSF4 has a crucial part in the de-nucleation of lens fibre cells through regulating DNase 2β expression level and DNase activity.28 Merath et al29 have demonstrated that the functional loss associated with autosomal-recessive HSF4 mutations is due to a loss of regulatory domains situated at the C-terminal end, indicating that the transcriptional activation of HSF4 is mediated by interactions between activator and repressor domains within the C-terminal end. Furthermore, the Hsf4 knockout mice, which demonstrate a cataract phenotype, have abnormal lens development with nondegenerated nuclei in the secondary fibre cells, suggesting a critical role of HSF4 in the lens fibre cell differentiation.30

References
Gilbert C, Foster A . Childhood blindness in the context of VISION 2020—the right to sight. Bull World Health Organ 2001; 79: 227–232.
Reddy MA, Francis PJ, Berry V, Bhattacharya SS, Moore AT . Molecular genetic basis of inherited cataract and associated phenotypes. Surv Ophthalmol 2004; 49: 300–315.
Krumpaszky HG, Klauss V . Epidemiology of blindness and eye disease. Ophthalmologica 1996; 210: 1–84.
Churchill A, Graw J . Clinical and experimental advances in congenital and paediatric cataracts. Philos Trans R Soc Lond B Biol Sci 2011; 366: 1234–1249.
Ionides A, Francis P, Berry V, Mackay D, Bhattacharya S, Shiels A et al. Clinical and genetic heterogeneity in autosomal dominant cataract. Br J Ophthalmol 1999; 83: 802–808.
Francis PJ, Berry V, Bhattacharya SS, Moore AT . The genetics of childhood cataract. J Med Genet 2000; 37: 481–488.
Shiels A, Bennett TM, Hejtmancik JF . Cat-Map: putting cataract on the map. Mol Vis 2010; 16: 2007–2015.
Berry V, Gregory-Evans C, Emmett W, Waseem N, Raby J, Prescott D et al. Wolfram gene (WFS1) mutation causes autosomal dominant congenital nuclear cataract in humans. Eur J Hum Genet 2013; 21: 1356–1360.
Pontikos N, Yu J, Moghul I, Withington L, Blanco-Kelly F, Vulliamy T et al. Phenopolis: an open platform for harmonization and analysis of genetic and phenotypic data. Bioinformatics 2017; 33 (15): 2421–2423.
McKenna A, Hanna M, Banks E, Svachenko A, Cibulskis K, Kernytsky A et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010; 20: 1297–1303.
McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GRS, Thormann A et al. The Ensembl variant effect predictor. Genome Biol 2016; 17: 122.
Glusman G, Caballero J, Mauldin DE, Hood L, Roach JC . Kaviar: an accessible system for testing SNV novelty. Bioinformatics 2011; 27: 3216–3217.
Köhler S, Vasilevsky NA, Engelstad M, Foster E, McMurry J, Aymé S et al. The Human Phenotype Ontology in 2017. Nucleic Acids Res 2016; 45 (D1): D865–D876.
Hamosh A, Scott AF, Amberger JS, Bocchini CA, McKusick VA . Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res 2005; 33: D514–D517.
Schwede T, Kopp J, Guex N, Peitsch MC . SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res 2003; 31: 3381–3385.
Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H et al. The Protein Data Bank. Nucleic Acids Res 2000; 28: 235–242.
Neudegger T, Verghese J, Hayer-Hartl M, Hartl FU, Bracher A et al. Structure of human heat-shock transcription factor 1 in complex with DNA. Nat Struct Mol Biol 2016; 23: 140–146.
Fujimoto M, Izu H, Seki K, Fukuda K, Nishida T, Yamada S-I et al. HSF4 is required for normal cell growth and differentiation during mouse lens development. EMBO J 2004; 23: 4297–4306.
Nakai A, Tanabe M, Kawazoe Y, Inazawa J, Morimoto RI, Nagata K . HSF4, a new member of the human heat shock factor family which lacks properties of a transcriptional activator. Mol Cell Biol 1997; 17: 469–481.
Tanabe M, Sasai N, Nagata K, Liu XD, Liu PC, Thiele DJ et al. The mammalian HSF4 gene generates both an activator and a repressor of heat shock genes by alternative splicing. J Biol Chem 1999; 274: 27845–27856.
Bu L, Jin Y, Shi Y, Chu R, Ban A, Eiberg H et al. Mutant DNA-binding domain of HSF4 is associated with autosomal dominant lamellar and Marner cataract. Nat Genet 2002; 31: 276–278.
Enoki Y, Mukoda Y, Furutani C, Sakurai H . DNA-binding and transcriptional activities of human HSF4 containing mutations that associate with congenital and age-related cataracts. Biochim Biophys Acta 2010; 1802: 749–753.
Sajjad N, Goebel I, Kakar N, Cheema AM, Kubisch C, Ahmad J . A novel HSF4 gene mutation (p.R405X) causing autosomal recessive congenital cataracts in a large consanguineous family from Pakistan. BMC Med Genet 2008; 9 (99).
Smaoui N, Beltaief O, BenHamed S, M’Rad R, Maazoul F, Ouertani A et al. A homozygous splice mutation in the HSF4 gene is associated with an autosomal recessive congenital cataract. Invest Ophthalmol Vis Sci 2004; 45: 2716–2721.
Behnam M, Imagawa E, Chaleshtori ARS, Ronasian F, Salehi M, Miyake N et al. A novel homozygous mutation in HSF4 causing autosomal recessive congenital cataract. J Hum Genet 2016; 61: 177–179.
Gillespie RL, O’Sullivan J, Ashworth J, Bhaskar S, Williams S, Biswas S et al. Personalized diagnosis and management of congenital cataract by next-generation sequencing. Ophthalmology 2014; 121 (2124–37): e1–e2.
Nakai A . Molecular basis of HSF regulation. Nat Struct Mol Biol 2016; 23: 93–95.
Cui X, Wang L, Zhang J, Du R, Liao S, Li D et al. HSF4 regulates DLAD expression and promotes lens de-nucleation. Biochim Biophys Acta 2013; 1832: 1167–1172.
Merath K, Ronchetti A, Sidjanin DJ . Functional analysis of HSF4 mutations found in patients with autosomal recessive congenital cataracts. Invest Ophthalmol Vis Sci 2013; 54: 6646–6654.
Shi X, Cui B, Wang Z, Weng L, Xu Z, Ma J et al. Removal of Hsf4 leads to cataract development in mice through down-regulation of gamma S-crystallin and Bfsp expression. BMC Mol Biol 2009; 10: 10.
Lv H, Huang C, Zhang J, Liu Z, Zhang Z, Xu H et al. A novel HSF4 gene mutation causes autosomal-dominant cataracts in a Chinese family. G3 2014; 4: 823–828.
Li D, Wang S, Ye H, Tang Y, Qiu X, Fan Q et al. Distribution of gene mutations in sporadic congenital cataract in a Han Chinese population. Mol Vis 2016; 22: 589–598.
Shi Y, Shi X, Jin Y, Miao A, Bu L, He J et al. Mutation screening of HSF4 in 150 age-related cataract patients. Mol Vis 2008; 14: 1850–1855.
Ke T, Wang QK, Ji B, Wang X, Liu P, Zhang X et al. Novel HSF4 mutation causes congenital total white cataract in a Chinese family. Am J Ophthalmol 2006; 142: 298–303.
Liu L, Zhang Q, Zhou L-X, Tang Z-H . A novel HSF4 mutation in a Chinese family with autosomal dominant congenital cataract. J Huazhong Univ Sci Technol Med Sci 2015; 35: 316–318.
Hansen L, Mikkelsen A, Nürnberg P, Nürnberg G, Anjum I, Eiberg H et al. Comprehensive mutational screening in a cohort of Danish families with hereditary congenital cataract. Invest Ophthalmol Vis Sci 2009; 50: 3291–3303.
Forshew T, Johnson CA, Khaliq S, Pasha S, Willis C, Abbasi R et al. Locus heterogeneity in autosomal recessive congenital cataracts: linkage to 9q and germline HSF4 mutations. Hum Genet 2005; 117: 452–459.
Acknowledgements
This work was supported by grants from the National Institute for Health Research Biomedical Research Centre at Moorfields Eye Hospital National Health Service Foundation Trust and UCL Institute of Ophthalmology (UK), Moorfields Eye Hospital Special Trustees (UK), Moorfields Eye Charity (UK), and the Foundation Fighting Blindness (USA). MM is supported by an FFB Career Development Award. We thank the members of the family for taking part in this study. We also thank Manav Ponnekanti who helped in the formatting of the document.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Berry, V., Pontikos, N., Moore, A. et al. A novel missense mutation in HSF4 causes autosomal-dominant congenital lamellar cataract in a British family. Eye 32, 806–812 (2018). https://doi.org/10.1038/eye.2017.268
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/eye.2017.268
This article is cited by
-
A novel missense mutation in the HSF4 gene of giant pandas with senile congenital cataracts
Scientific Reports (2021)
-
GJA8 missense mutation disrupts hemichannels and induces cell apoptosis in human lens epithelial cells
Scientific Reports (2019)
-
Genetic landscape of isolated pediatric cataracts: extreme heterogeneity and variable inheritance patterns within genes
Human Genetics (2019)
-
Whole-genome sequencing reveals a recurrent missense mutation in the Connexin 46 (GJA3) gene causing autosomal-dominant lamellar cataract
Eye (2018)
