
Abstract
Von Hippel–Lindau disease (vHL) is a rare hereditary tumour predisposition with multiorgan involvement that is not always easily recognized. The disease is reported to be almost fully penetrant at age 60 years. Previous estimates of vHL prevalence and incidence are all regional and vary widely. Most are >20 years old and prone to selection bias because of inclusion of only clinically affected vHL patients who were diagnosed before genetic testing was available. In an unselected cohort of all known Danish carriers of a disease-causing VHL variant, we assessed vHL penetrance on a national basis. We further used national health registers to identify individuals who fulfilled the clinical diagnostic vHL criteria based on their registered diagnostic codes, but had not been diagnosed with vHL. We also assessed the medical histories of first-degree relatives to identify familial cases. This study gives the first national estimates of vHL prevalence (1 in 46 900 individuals) and birth incidence (1 in 27 300 live births). vHL has been underdiagnosed in Denmark, and as many as 25% of the overall vHL cohort (diagnosed+undiagnosed patients) have a missed diagnosis in spite of fulfilling the international diagnostic criteria. We found an overall penetrance of 87% at age 60 years. When considering only vHL patients who have not attended surveillance, 20% will still be asymptomatic at age 60 years. This should be considered in the context of genetic counselling, especially when assessing the risk of vHL in asymptomatic adult first-degree relatives who are often not genetically tested.
Similar content being viewed by others


Introduction
Von Hippel–Lindau disease (vHL) (OMIM number 193300) is a hereditary autosomal dominant tumour predisposition in which carriers of a disease-causing germline variant in the VHL gene are at risk of developing benign and malignant tumours in multiple organs.1 The VHL gene is a tumour suppressor gene that plays a vital role in the regulation of angiogenesis and cell division.1 The disease is reported to be highly penetrant with almost 100% of patients affected at age 60 years.2, 3 The most common manifestations are benign haemangioblastomas that can develop throughout the CNS, typically in the cerebellum, spinal cord, and retina. Patients are also at risk of renal cell carcinoma (RCC), pheochromocytoma, pancreatic neuroendocrine tumours (PNETs), endolymphatic sac tumours (ELSTs), as well as cysts in the kidneys, pancreas, and epididymis or broad uterine ligament.1 No systematic treatment exists, and the best available treatment option is surgical tumour removal before major sequelae occur. vHL patients are advised to undergo prophylactic surveillance to facilitate early tumour diagnosis.1, 4 The prevalence of vHL has been reported to be between 1 in 39 000 and 1 in 91 000 individuals in different regional populations,2, 3, 5, 6 but has never been investigated on a national basis. Based on previous estimates, in the Danish population of ∼5.6 million residents (http://www.statistikbanken.dk/statbank5a/default.asp?w=1920, accessed 1-6-2016), we would expect between 65 and 140 individuals to have the diagnosis. The disease has most likely been underdiagnosed in Denmark, as only 59 living carriers of a disease-causing VHL variant were known in 2008.7 vHL is normally diagnosed on a genetic basis, frequently presymptomatic in familial cases. However, diagnosis of isolated vHL cases is often delayed compared with familial cases,8 and may never be recognized, as a clinical diagnosis requires at least two manifestations, often involving multiple medical specialists.4 A timely diagnosis is essential to establish surveillance and offer genetic counselling to at-risk family members. We aimed to determine the prevalence, birth incidence, and penetrance of vHL in a national study.
Subjects and methods
We performed a retrospective cohort study using the national vHL Research Database and national health registers. For this part of the study, we included individuals who had been alive at some point between 1 January 1977 and 10 December 2015. In this period, the data registered about Danish residents in the national health registers was assessed to be accurate and complete because of mandatory registration of all hospital admissions from 1977.
Categories identified for individuals
We identified individuals belonging to the following below mentioned categories.
Probands
Diagnosed vHL patients
We obtained consent from 87% (106 of 122) of patients who were known through the vHL Research Database to be diagnosed with vHL and alive during the study period. Their clinical characteristics were evaluated through interviews and hospital records. For an overview of the inclusion process, see Figure 1. Two subgroups of diagnosed patients were defined:

Flowchart showing the inclusion process of the diagnosed vHL population.
(1) Mutation-positive patients (N =76): carriers of disease-causing VHL germline variants (either molecularly confirmed or obligate variant carriers), or (2) Mutation-negative patients (N =8): individuals who fulfil the clinical diagnostic vHL criteria,1, 4 but in whom no disease-causing VHL germline variant could be identified.
The VHL variants identified in the cohort of diagnosed vHL patients have previously been described by others.9
Assumed vHL patients
Individuals who fulfilled the clinical diagnostic vHL criteria based on their registered diagnostic codes in the Patient Register, the Cancer Register, the Causes of Death register, and the Pathology Register. These individuals had not yet been diagnosed with vHL. Our search of the national health registers is thoroughly described in the Supplementary Subjects and methods. We used the international clinical diagnostic criteria for optimal comparison with other studies. According to the Danish diagnostic criteria, an individual with a negative family history will fulfil the diagnostic criteria by having any two vHL-related manifestations, and there is no requirement for at least one to be a haemangioblastoma as there is in the international criteria.1 The differences between international and Danish criteria are outlined in Table 1. Based on the different diagnostic criteria we defined two subgroups of assumed vHL patients (their clinical characteristics are described in detail in Supplementary Table 3):
-
a)
Assumed patient (International) (N =49): individuals fulfilling the international clinical diagnostic criteria by having at least one haemangioblastoma and one other vHL-related manifestation or multiple haemangioblastomas as registered in the health registers. These individuals also fulfil the Danish criteria.
-
b)
Assumed patient (Danish) (N =68): individuals fulfilling the Danish clinical diagnostic criteria by having at least two vHL-related manifestation of any type registered in the health registers.
To assess whether the probands had familial or nonfamilial vHL, secondary cases were identified among first-degree relatives:
First-degree relatives to the 122 diagnosed vHL patients ( N =256)
Parents, siblings, and children to diagnosed vHL patients who had been alive at some point during the study period were identified through the vHL Research Database. They either had a negative genetic analysis (N=89) or no known vHL-related symptoms (N=167). We were able to assess the medical histories of 135 of the 167 (83%) first-degree relatives as registered in the national health registers.
We found registrations of a vHL-related manifestation in three deceased relatives who had not previously been diagnosed (the pedigrees are shown in Supplementary Figure 3A and B where the families are numbered vHL family 1, 2, and 3). One individual had kidney cancer at age 74 years (pathology unspecified) and had a parent and siblings with vHL (vHL family 1), whereas two individuals had adenocarcinoma of the pancreas at ages 50 and 52 years, one had a child with vHL (vHL family 2), and the other had a sibling with vHL (vHL family 3). These three were included as diagnosed vHL patients in the prevalence and incidence analyses.
First-degree relatives to the 68 assumed vHL patients ( N =331)
Parents, siblings, and children to the assumed vHL patients, identified through the Civil Registration System, and in some cases also Church records, and Population Censuses. We evaluated the full medical histories as registered in the national health registers of 205 of the 331 (62%) first-degree relatives, and identified three relatives with at least one vHL-related manifestation (three familial cases); their pedigrees are shown in Supplementary Figure 4.
In Table 2, the characteristics of the assumed patients are compared with the diagnosed patients who would fulfil the Danish diagnostic criteria based on their phenotype alone.
Estimation of vHL penetrance
vHL penetrance was estimated based on 150 diagnosed vHL patients. We did not use information from the national health registers for the penetrance analysis, and therefore included all diagnosed vHL patients for whom we had information about their first diagnosis from the vHL Research Database, regardless of when they had been alive. Their first manifestations had been diagnosed between 1878 and 2014. We used Kaplan–Meier curves to calculate their age-related cumulative incidence of the diagnosis of (A) their first manifestation (regardless of whether it asymptomatic or symptomatic at diagnosis) and (B) their first symptomatic manifestation. Individual were included from birth until their first manifestation diagnosis. Individuals were censored at the time of death, and in model B also at the time of their first asymptomatic manifestation diagnosis. The penetrance of first symptomatic manifestations was used to estimate the natural history of the disease in individuals not undergoing surveillance. Statistical analysis was performed using R, version 3.2.5 with the package survival (version 2.28-3, Lucent Technologies, Murray Hill, NJ, USA).
Point prevalence and birth incidence
The prevalence and incidence estimates were calculated based on the international diagnostic criteria and separately based on the Danish diagnostic criteria. The point prevalence was calculated on 1 January 2014 as the proportion of all living diagnosed vHL patients AND assumed vHL patients in the Danish population. The birth incidence was estimated by calculating the diagnosed vHL patients AND the assumed vHL patients born in the 20-year period from 1945 through 1964 as a proportion of the total number of live births in the same period. This 20-year period was chosen as we expected the ascertainment of assumed vHL patients to be most complete for this period. All possible vHL patients would be younger than 32 years in 1977 when the national registration of hospital admissions started. Most would have been alive at this age, and almost 60% would not yet have had their first symptomatic vHL manifestations if they did not attend vHL surveillance. The included patients born before 1964 would be at least 50 years at the end of the registration period in 2015. At this age, the majority (78%) of the vHL patients would have had symptomatic manifestations, if they did not attend surveillance. We obtained information about the Danish population size on 1 January 2014 and the number of live births from 1945 to 1964 from Statistics Denmark (http://www.statistikbanken.dk/statbank5a/default.asp?w=1920).
Ethics
The study was approved by the Danish Regional Committees on Biomedical Ethics (journal no. H-2-2010-012) and the Danish Data Protection Agency (journal no. 2009-41-3994).
Results
Estimation of vHL penetrance
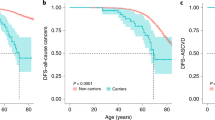
Of all diagnosed vHL patients, 87% (130 of 150) had manifestations. Of these, 83 had a symptomatic first manifestation, whereas it was asymptomatic in 38 individuals (diagnosed because of surveillance or at autopsy). We did not know whether the first manifestations of nine deceased individuals had been symptomatic or asymptomatic. As all nine were diagnosed before surveillance recommendations were routinely used, we included them as being symptomatic. If all nine were instead assumed to be asymptomatic, the penetrance estimates were similar (Supplementary Figure 5). The age-related penetrance for all first manifestations as well as for manifestations diagnosed because of symptoms alone are shown in Table 3 and illustrated in Figure 2.

Estimated vHL penetrance (N=150). A: age-related cumulative incidence of all first vHL manifestations. B: age-related cumulative incidence of all symptomatic first vHL manifestations. In A, all first manifestations (symptomatic and asymptomatic) were considered as events. Subjects are censored at age of death or study end, if no first manifestation has been diagnosed. In B, only symptomatic first manifestations are considered, whereas patients with first asymptomatic manifestations were censored at the age at diagnosis. In this analysis, the nine first manifestations with unknown mode of diagnosis were assumed to be symptomatic. The estimates for 70 years were not included in the table, as the number of included individuals at this age is too small for a reliable estimate.
Evaluation of the search strategy
The strategy used to identify the assumed vHL patients in the search of the national health registers missed 17% (21 of 122) of the diagnosed vHL patients who had been alive between 1977 and 2015 and were therefore expected to have vHL-related diagnostic codes registered in the national health registers. Their clinical characteristics are described in Supplementary Table 4. Six had no diagnosed manifestations, three had multiple RCC tumours, five had only one diagnosed manifestation, and none had the diagnostic code for vHL, DQ858D, that was used in the register search.
Point prevalence and birth incidence
We estimated the point prevalence and birth incidence of vHL (Table 4) based on:
-
a)
The international clinical diagnostic criteria:1 all possible vHL patients according to the international criteria (all diagnosed patients and those of the assumed patients with at least one haemangioblastoma), and
-
b)
The Danish clinical diagnostic criteria:4 all possible vHL patients (all diagnosed and assumed patients). Both estimates of assumed vHL patients were further adjusted for the approximated degree of uncertainty (we added 17% as a measure of missed patients in the search).
Discussion
This study gives the first national estimates of vHL prevalence and birth incidence. Ninety-one living individuals had diagnosed vHL, whereas ∼29 living individuals (25 assumed vHL patients+17%) were estimated to have a missed vHL diagnosis in spite of fulfilling the international clinical diagnostic criteria based on their registered diagnostic codes. Previous studies of vHL prevalence and incidence have been regional and based on cohorts of clinically affected patients who were diagnosed before accurate genetic testing was available.2, 3, 5, 10 Regional studies may be influenced by founder effects and skewed distribution of affected families, although inclusion on a clinical basis gives rise to selection bias, when asymptomatic VHL-variant carriers are missed. Furthermore, most previous studies included selected cohorts of vHL patients referred by specialists or treated at specific hospitals, and this might explain the large discrepancies between the estimates.2, 3, 5, 10 Our estimated prevalence of ∼1 in 46 900 is lower than the highest estimate of 1 in 39 000 individuals reported in Freiburg.5 In this region, the founder variant, c.292T>C is prevalent.11, 12 The variant is associated with a relatively benign natural history that has probably facilitated its vast spread in several geographic areas resulting in a higher regional prevalence.11, 12 In contrast, our prevalence and incidence estimates were both higher than reported by Maher et al2 in 1990 in East Anglia (prevalence: 1 in 53 000 and incidence: 1 in 36 000 births) and by Maddock et al.3 in 1994 in North West England (prevalence: 1 in 85 000 and incidence: 1 in 45 500 births). No founder effects have been found in England or Denmark.9, 13 In 2008, the prevalence and incidence was updated for the same region as studied by Maddock et al,10 and were slightly lower than 14 years earlier (prevalence: 1 in 91 000 and the birth incidence of ∼1 in 53 000 births), even though patients were included on a clinical and genetic basis.10 The disparity is most likely because of methodological differences. Maddock et al3, 10 took estimates of yet undiagnosed variant carriers into account, whereas the new study counted only diagnosed patients.
We found an overall penetrance of 87% at age 60 years in a national cohort including >90% of all diagnosed vHL patients. When only considering patients who had not attended surveillance before their first diagnosis, 20% would still be asymptomatic at age 60 years. This was remarkably lower than previous reports of almost full penetrance (99% and 98.5%) at age 60 years.2, 3 The phenotypic spectrum of our diagnosed vHL cohort is wider than in other reported cohorts because of an unselected patient inclusion. In our cohort, 14% (15 of 106) were asymptomatic or obligate VHL variant carriers without manifestations. In two previous studies, only 1 out of 236 (0.5%) and 3 out of 83 (3.6%) were asymptomatic obligate variant carriers, respectively, and the rest were clinically affected.2, 3 This suggests a bias towards inclusion of patients with severe phenotypes and an underrepresentation of those mildly affected.
In our prevalence and incidence analyses, we used the international diagnostic vHL criteria for direct comparisons with previous estimates. Nevertheless, the international criteria would have missed as many as 13% of the diagnosed VHL variant carriers who have only visceral lesions and no haemangioblastomas at the time of their second manifestation diagnosis, when they would have fulfilled the diagnostic criteria on a purely clinical basis (Table 1). This underlines the usefulness of the more inclusive Danish diagnostic criteria compared with the international criteria.
In practice, the diagnosis of vHL in a clinical setting depends on the individual family and takes multiple aspects, especially differential diagnosis and comprehensive genetic testing into account. The greatest challenge lies in assessing whether mutation-negative patients with only visceral lesions, in whom other differential diagnoses have been ruled out, should attend vHL surveillance, as they will still fulfil our diagnostic criteria. These cases require careful assessment and thorough investigation of possible differential diagnosis before the vHL diagnosis is given, as it entails comprehensive surveillance recommendations for both the proband and at-risk first-degree relatives.4
The assumed vHL patients were identified in a register search and not all have yet been systematically genetically tested. The likelihood of them truly having vHL was evaluated in comparison with the diagnosed vHL patients (Table 1). The most common first manifestation type was CNS haemangioblastomas in both groups, and the second most frequent for the assumed patients were paraganglioma and pheochromocytoma that were rarely the first manifestation for the diagnosed vHL patients. Approximately 30% of the assumed patients had only visceral lesions at the time of their second manifestations. The most frequent combination (in 60%, 12 of 20) was a neuroendocrine tumour (a pheochromocytoma, a paraganglioma, or a PNET) along with RCC. Some of these individuals may represent phenocopies who by chance have a rare endocrine tumour and later in life develop sporadic RCC. The mean age at RCC diagnosis among our assumed vHL patients was 61 years (range: 40–85 years, N=21). This corresponds better with the generally older age at debut in sporadic RCCs cases,14 and is higher than the mean ages at RCC onset for vHL patients of 40–45 years (N=247, range: 20–69 years).4 On the other hand, as many as 11% of patients with an apparently isolated pheochromocytoma have a germline VHL variant.15 Other differential diagnosis should be considered along with vHL in the initial assessment. Overall, seven of the assumed patients had a combination of pheochromocytoma, paraganglioma, and/or pancreatic tumours. Three of them had a total of five first-degree relatives with pheochromocytoma and/or paraganglioma without any other vHL-related lesions, making multiple endocrine neoplasia 2A, SDHx-, FH-, MAX-, or TMEM127- associated hereditary pheochromocytoma/paraganglioma relevant differential diagnoses.16
The majority of the assumed cases were nonfamilial based on their families’ medical histories alone. This is not surprising, as the possibility of vHL is easier to recognize when several family members are affected. Only a tenth of the diagnosed vHL patients had no family history, and others have reported negative family histories in between 9 and 23% of vHL patients.3, 13, 17, 18 Estimates of de novo vHL cases have been reported in between 3 and 21%.13, 17, 19 Among our diagnosed vHL patients, we estimated that the disease was caused by de novo variants in 21% (22 of 106) of the patients. Our assessment was, however, based on pedigree analyses and genetically confirmed in only one family. In the rest, the proband’s parents had not been genetically tested, and could represent asymptomatic or mildly affected variant carriers, or germline mosaicism.13, 18
Even though >20% (15 of 71) of the assumed vHL patients identified in this study had undergone genetic testing (VHL sequencing and either Southern blotting or multiplex ligation-dependent probe amplification – depending on when the patient had been tested), no disease-causing VHL variants had been found, not even in patients with a pathologically confirmed CNS haemangioblastoma (22% (11 of 49) had been tested). Previous reports suggest that up to 15% of patients fulfilling the diagnostic criteria have no identifiable VHL variants.17, 20, 21 Some might be phenocopies, mosaics, or have alterations in other genes that give rise to a vHL-like phenotype. Better knowledge about the risk of additional manifestations in such families is needed to facilitate appropriate surveillance measures.
The Danish national health registers provide thorough documentation of all health care system contacts throughout each resident’s lifetime. The use of multiple health registers over an observation period of almost 40 years enabled us to achieve a high degree of ascertainment of vHL families on a national basis. Nevertheless, our method could still have underestimated the number of assumed vHL patients, as the search had certain limitations. (1) Individuals with multiple manifestations of the same type (other than haemangioblastomas) may have been missed. These individuals fulfil the Danish diagnostic criteria.4 However, all patients fulfilling the international criteria were identified, as we included all patients with multiple haemangioblastomas. The estimates of prevalence and incidence based on the international criteria are expected to be accurate. (2) The diagnostic codes are registered by the treating physicians, giving rise to nonuniform coding strategies throughout the country. (3) We may have missed individuals who had manifestations diagnosed or who died before 1977 when the study period started. Contrary, our group of assumed vHL patients includes 23 individuals with unspecific diagnostic codes for at least one registered manifestation, and some may represent non-vHL-related lesions that would instead overestimate the vHL frequency.
Of the vHL patients, 87% will have had their first manifestation diagnosed at age 60 years. As many as 20% of the predisposed individuals who had not attended surveillance did not have had any vHL-related symptoms of the disease at 60 years of age. This should be considered in the context of genetic counselling, especially when assessing the risk of vHL in asymptomatic adult first-degree relatives who are often not genetically tested.
References
Maher ER, Neumann HP, Richard S : von Hippel-Lindau disease: a clinical and scientific review. Eur J Hum Genet 2011; 19: 617–623.
Maher ER, Iselius L, Yates JR et al: Von Hippel-Lindau disease: a genetic study. J Med Genet 1991; 28: 443–447.
Maddock IR, Moran A, Maher ER et al: A genetic register for von Hippel-Lindau disease. J Med Genet 1996; 33: 120–127.
Binderup ML, Bisgaard ML, Harbud V et al: Von Hippel-Lindau disease (vHL). National clinical guideline for diagnosis and surveillance in Denmark. 3rd edition. Dan Med J 2013; 60: B4763.
Neumann HP, Wiestler OD : Clustering of features of von Hippel-Lindau syndrome: evidence for a complex genetic locus. Lancet 1991; 337: 1052–1054.
Wilding A, Ingham SL, Lalloo F et al: Life expectancy in hereditary cancer predisposing diseases: an observational study. J Med Genet 2012; 49: 264–269.
Poulsen ML, Budtz-Jorgensen E, Bisgaard ML : Surveillance in von Hippel-Lindau disease (vHL). Clin Genet 2010; 77: 49–59.
Hes FJ, van der Luijt RB, Janssen AL et al: Frequency of Von Hippel-Lindau germline mutations in classic and non-classic Von Hippel-Lindau disease identified by DNA sequencing, Southern blot analysis and multiplex ligation-dependent probe amplification. Clin Genet 2007; 72: 122–129.
Dandanell M, Friis-Hansen L, Sunde L, Nielsen FC, Hansen TV : Identification of 3 novel VHL germ-line mutations in Danish VHL patients. BMC Med Genet 2012; 13: 54.
Evans DG, Howard E, Giblin C et al: Birth incidence and prevalence of tumor-prone syndromes: estimates from a UK family genetic register service. Am J Med Genet A 2010; 152A: 327–332.
Brauch H, Kishida T, Glavac D et al: Von Hippel-Lindau (VHL) disease with pheochromocytoma in the Black Forest region of Germany: evidence for a founder effect. Hum Genet 1995; 95: 551–556.
Bender BU, Eng C, Olschewski M et al: VHL c.505T>C mutation confers a high age related penetrance but no increased overall mortality. J Med Genet 2001; 38: 508–514.
Richards FM, Payne SJ, Zbar B, Affara NA, Ferguson-Smith MA, Maher ER : Molecular analysis of de novo germline mutations in the von Hippel-Lindau disease gene. Hum Mol Genet 1995; 4: 2139–2143.
Maher ER : Genetics of familial renal cancers. Nephron Exp Nephrol 2011; 118: e21–e26.
Neumann HP, Bausch B, McWhinney SR et al: Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med 2002; 346: 1459–1466.
Jafri M, Maher ER : The genetics of phaeochromocytoma: using clinical features to guide genetic testing. Eur J Endocrinol 2012; 166: 151–158.
Cybulski C, Krzystolik K, Murgia A et al: Germline mutations in the von Hippel-Lindau (VHL) gene in patients from Poland: disease presentation in patients with deletions of the entire VHL gene. J Med Genet 2002; 39: E38.
Sgambati MT, Stolle C, Choyke PL et al: Mosaicism in von Hippel-Lindau disease: lessons from kindreds with germline mutations identified in offspring with mosaic parents. Am J Hum Genet 2000; 66: 84–91.
Nordstrom-O'Brien M, van der Luijt RB, van RE et al: Genetic analysis of von Hippel-Lindau disease. Hum Mutat 2010; 31: 521–537.
Rasmussen A, Nava-Salazar S, Yescas P et al: Von Hippel-Lindau disease germline mutations in Mexican patients with cerebellar hemangioblastoma. J Neurosurg 2006; 104: 389–394.
Glasker S, Bender BU, Apel TW et al: The impact of molecular genetic analysis of the VHL gene in patients with haemangioblastomas of the central nervous system. J Neurol Neurosurg Psychiatry 1999; 67: 758–762.
Acknowledgements
The study was financially supported by the Danish Cancer Society, the Lundbeck Foundation, and the Danish association of von Hippel–Lindau patients and their relatives. The funding organizations solely provided financial support and had no influence on the study design, data collection or analysis, or the decision to publish. We thank the participating vHL families. We are grateful to Steen Kølvraa who helped plan the study register search, but passed away before the study end. We also thank the Danish vHL coordination group for their collaboration; Kaj Albrechtsen, Anette Møller Jensen, and Thomas Rosenberg for identifying the first vHL families; Marie Louise Dylov and Elizabeth Aagren Mikkelsen who helped draw pedigrees; and Anne-Sophie Stendell who evaluated hospital records for individuals with registered retinal ICD codes.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Binderup, M., Galanakis, M., Budtz-Jørgensen, E. et al. Prevalence, birth incidence, and penetrance of von Hippel–Lindau disease (vHL) in Denmark. Eur J Hum Genet 25, 301–307 (2017). https://doi.org/10.1038/ejhg.2016.173
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2016.173
This article is cited by
-
Epidemiology and economic burden of Von Hippel-Lindau Disease-associated central nervous system hemangioblastomas and pancreatic neuroendocrine tumors in the United States
Orphanet Journal of Rare Diseases (2024)
-
Targeting HIF-2 Alpha in Renal Cell Carcinoma
Current Treatment Options in Oncology (2023)
-
VHL mosaicism: the added value of multi-tissue analysis
npj Genomic Medicine (2022)
-
The incidence of consecutive manifestations in Von Hippel-Lindau disease
Familial Cancer (2019)
