
Abstract
As an example of optimizing population-specific genotyping assays using a whole-genome sequence reference set, we detail the approach that followed to design the Axiom-NL array which is characterized by an improved imputation backbone based on the Genome of the Netherlands (GoNL) reference sequence and, compared with earlier arrays, a more comprehensive inclusion of SNPs on chromosomes X, Y, and the mitochondria. Common variants on the array were selected to be compatible with the Illumina Psych Array and the Affymetrix UK Biobank Axiom array. About 3.5% of the array (23 977 markers) represents SNPs from the GWAS catalog, including SNPs at FTO, APOE, Ion-channels, killer-cell immunoglobulin-like receptors, and HLA. Around 26 000 markers associated with common psychiatric disorders are included, as well as 6705 markers suggested to be associated with fertility and twinning. The platform can thus be used for risk profiling, detection of new variants, as well as ancestry determination. Results of coverage tests in 249 unrelated subjects with GoNL-based sequence data show that after imputation with 1000G as a reference, the median concordance between original and imputed genotypes is above 98%. The median imputation quality R2 for MAF thresholds of 0.001, 0.01, 0.05, and >0.05 are 0.05, 0.28, 0.80, 0.99, respectively, for the 1000G imputed SNPs, with a similar quality for the autosomes and X chromosome, showing a good genome-wide coverage for association studies after imputation.
Similar content being viewed by others


Introduction
Genome-wide association studies (GWAS) in large population samples have been the key method to identify genetic variants involved in complex human traits.1, 2 Multiple successful GWAS studies have been reported ranging from body size, metabolomics, and medically relevant traits (reviewed),3, 4 to hormones,5 personality,6 educational attainment,7 and lifestyle characteristics.8, 9, 10
The major technology behind these successes is the relatively cheap genotyping, in comparison with full genome sequencing, of DNA samples on genotyping arrays with 300 K–5 M single-nucleotide polymorphisms (SNPs), followed by imputation of the unmeasured SNPs. Initially, the contents of these arrays were determined by the manufacturers, but recently companies also allow researchers to select the variants on an array. Here, we focus on the Axiom array, a genotyping solution from Affymetrix, Inc., which provides a high throughput platform for high-density SNP genotyping on a diverse range of sample types. This array has been used for several large population-wide genome-screening projects including the UK biobank11 and the GERA cohorts.12 We describe a similar custom-made Axiom array for the Netherlands population, the Axiom-NL that allows good imputation and enhances association, and risk score analysis on DNA samples collected within Dutch Biobanks, such as the large number of Biobanks collaborating in BBMRI-NL.13 Notwithstanding the application to this specific population, the SNP selection procedures and coverage testing can provide a general guideline for customizing the Axiom array in other populations for which valid reference sequence genomes are available.
Materials and methods
SNP selection for the Axiom-NL array
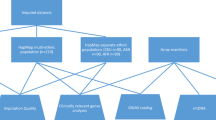
An overview of the SNP selection is provided in Figure 1, a stepwise procedure of the selection is given in the Supplementary materials. The core of the array was optimized for genome-wide coverage using genotype imputation. The Affymetrix SNP 6.0 array formed the starting point for selection of markers.14 SNPs were selected that passed quality control (see supplementary methods), including if the replicate genotype error rate in control samples was <1% in prior experiments, and provided the most tagging information. We then selected up to 10 additional markers per mega base in areas that had a low imputation quality (mean R2<0.35) based on imputations with these SNPs alone. Here, SNPs were selected only if they were present in the Dutch population based on the GoNL reference sequence data.13 We prioritized them based on the following criteria: R2<0.30, MAF>0.07, and preferably high LD r2>0.5–0.9 with other weakly imputed SNPs in the 1 MB region. Selected SNPs in high LD with each other were removed (PLINK 1.07 —indep 200 10 1.5).15 Applying a MAF 0.07 threshold here is crucial, because the LD pruning step otherwise selects only rare independent SNPs. The same SNP selection approach was used for chromosome X, to achieve similar coverage as the autosomes.

Axiom-NL marker selection and content. (a) Simplified flow chart depicting the strategy for selecting markers for Axiom-NL array. (b) A pie graph representing the broad breakdown of marker categories included in Axiom-NL.
In addition, laboratory validated SNPs on commercially available microarrays (eg, Affymetrix UK Biobank Axiom Array, Affymetrix Axiom Biobank Array used in the Million Veteran Program, and the Illumina (San Diego, CA, USA) Infinium Psych Array) were added as they are informative for several traits and disease studies. An important consideration for selection of SNPs from these platforms was to focus on selecting common SNPs (MAF>0.01), with the exclusion of important rare SNPs known to have associations with complex traits of interest (Supplementary Figure 1). Furthermore, chromosome Y markers were selected to be the same as the Axiom UK Biobank. Mitochondrial markers were selected from Axiom UK Biobank and the Human Mitochondrial Genome Database to represent the most frequent Dutch haplotypes. The final annotation file of the selected 671 222 SNPs is available for download at www.avera.org/axiom.
Estimating genome-wide concordance
To estimate the coverage of the Axiom-NL design, we used a two genome-wide reference approach, where SNPs from the GoNL reference were re-imputed with the 1000 genomes reference based on only the selected Axiom-NL SNPs. The GoNL reference data are 769 individuals, spread across the Netherlands that were sequenced, aligned to genome build 37, variant-called, and phased for imputation for chromosomes 1–22 and X.16 The 1000 Genomes reference panel are 2504 sequenced individuals from several populations worldwide.17
From the GoNL sequence data, 249 unrelated women were identified and their genotype data for chromosomes 1–22 and X were extracted (N=22 932 747 SNPs). Data from only women were selected to facilitate that chromosome X imputes similarly to the autosomes. From these data, the 671 222 SNPs that were present on the Axiom-NL array were extracted as input data for the 1000G imputation. These SNPs were filtered with the following criteria: minor allele frequency (MAF) <0.01, call rate <0.95, Hardy–Weinberg Equilibrium test P-value <10−5, and SNPs having the same alleles as in 1000G, leaving 618 889 SNPs. Strands were checked (SHAPEIT 2.7r790) and flipped (PLINK 1.07) if required before phasing. This set thus mimics a quality controlled pre-imputation genotyped Axiom-NL data set. For comparison the same procedure was applied for the SNP annotation lists from two similarly sized commercially available arrays, the Affymetrix Axiom Biobanking array and the Illumina Infinium OmniExpress-24 BeadChip. Subsequently, the three data sets were phased with SHAPEIT 2.7r790 and imputed against the 1000G all reference panel Phase 3 (October 2014) for the autosomes, and 1000G All Phase 1 interim (June 2011) for the X chromosome. Note that we could not use the GoNL or HRC as an imputation reference panel here, since the subjects from our to be imputed GoNL data set are present in these reference panels. As such they will be imputed back perfectly as their haplotypes would match 100% (previously tested). This would therefore not tell us anything about the ability to impute the genome from just the Axiom-NL SNP list. The imputations were done with IMPUTE2.3.1 using standard protocols.18 From the imputed data, best-guess genotypes were calculated for all 82 943 231 SNPs (Plink 1.90). For 12 205 845 overlapping SNPs between GoNL and 1000G with MAF>0 in the imputed data of all three sets and both references, the concordance between the GoNL sequence and the 1000G imputations was calculated with PLINK 1.90. For each SNP, polymorphic in all sets, the median, average, and SD was calculated for the imputation quality R2 (Quicktest 0.95) in MAF bins >0–0.001, >0.001–0.01, >0.01–0.05, and >0.05 (SPSS 22). This was done for the full 1000 genomes imputation and for the SNPs overlapping with GoNL.
Results
For the imputation quality R2, where zero is extremely sub-optimal and one is excellent, the results from the three platforms are presented in Table 1. For all 1000 genomes imputed SNPs, autosomes, and chromosome X, the median R2 values for the Axiom-NL platform are 0.0496 for MAF 0–0.001, 0.281 for MAF >0.001–0.01, 0.805 for MAF >0.01–0.05, and 0.991 for MAF >0.05 (Table 1). For chromosome X alone these values are 0.120, 0.555, 0.838, and 0.993, respectively, indicating that the rare chromosome X SNPs are imputed slightly better than the autosomes and the common SNPs equally well. With these results, the Axiom-NL platform is just in between the Affymetrix Axiom Biobanking array and the Illumina Infinium OmniExpress-24 BeadChip as shown in Table 1. The differences in imputation quality are, however, extremely small, between the three platforms for all MAFs and all chromosomes. When selecting SNPs that are present in the GoNL and the 1000G reference data, the true variants in the Dutch population, the results show an even better imputation quality. The main reason behind this is the large number of rare SNPs, which are likely absent in the Dutch population are now excluded, which improves the median and mean scores.
The concordance rates of the genotyped GoNL SNPs that were re-imputed with a 1000 Genomes imputation were generally high for most SNPs in the genome (Table 2). For the 12 205 845 markers with MAF>0, being polymorphic in the imputed data for all three platforms in the 249 women, up to 59.6% can be re-imputed at high quality. With a lower level of quality (down to 80% concordance), only 2.8% of the genome is not covered well. For the concordance measurements, the Axiom-NL array is again in between the Affymetrix Axiom Biobanking array and the Illumina Infinium OmniExpress-24 BeadChip, where the Illumina chip performs slightly better and the Axiom Biobanking array slightly worse. Genome wide, there are no large differences between the chips imputation quality.
Discussion
The Axiom-NL Array was developed with a custom backbone to provide optimal imputation for the Dutch population with an improvement of coverage for chromosome X. The design incorporates a significant clinical relevance focus by including the common variants from two large consortia (Psychiatric Genomics Consortium and UK Biobank). Over 60 000 markers are included from the UK BioBank array including known GWAS hits from the NHGRI GWAS catalog, with additional modules including apoE, HLA, cardiometabolic, and mitochondrial SNPs. For projects of interest to twin registers, 6705 additional candidate SNPs implicated in fertility and twinning were selected.
With a general reference set, and markers selected for the Axiom-NL array we can re-impute with high confidence, keeping in mind the exception that rare alleles (MAF<0.001) are never imputed well.19 Also the methods we utilized, having only used the sequence of 249 samples, the imputation and presence of minor alleles with MAF <0.01 was likely not optimal. However, for comparison tests this should not matter and the imputation is of similar quality to other commercially available chips namely the Affymetrix Axiom Biobanking array and the Illumina Infinium OmniExpress-24 BeadChip. Finally, our method tested the coverage using two reference data sets and the concordance between genotyped SNPs, and re-imputed SNPs inherently assumes that the SNPs need to be present in both reference data sets. As such we thus assume that population-specific SNPs, for example, present only in GoNL are covered and imputed just as well.
Knowledge generation in genetic epidemiology depends increasingly on the use of SNP-array based GWA studies, including (bivariate) GCTA and polygenic risk score analysis, or the combination of summary statistic information for multiple traits as in LD-score regression and Mendelian Randomisation.20 We here show that customized population-specific arrays for imputation-based GWA testing can be a valuable tool to generate high quality GWA results.
References
Stranger BE, Stahl EA, Raj T : Progress and promise of genome-wide association studies for human complex trait genetics. Genetics 2011; 187: 367–383.
Visscher PM, Brown MA, McCarthy MI, Yang J : Five years of GWAS discovery. Am J Hum Genet 2012; 90: 7–24.
Geschwind DH, Flint J : Genetics and genomics of psychiatric disease. Science 2015; 349: 1489–1494.
Chang CQ, Yesupriya A, Rowell JL et al: A systematic review of cancer GWAS and candidate gene meta-analyses reveals limited overlap but similar effect sizes. Eur J Hum Genet 2014; 22: 402–408.
Ruth KS, Campbell PJ, Chew S et al: Genome-wide association study with 1000 genomes imputation identifies signals for nine sex hormone-related phenotypes. Eur J Hum Genet 2016; 24: 284–290.
Genetics of Personality Consortium Genetics of Personality Consortium, de Moor MH Genetics of Personality Consortium, van den Berg SM et al: Meta-analysis of genome-wide association studies for neuroticism, and the polygenic association with major depressive disorder. JAMA Psychiatry 2015; 72: 642–650.
Rietveld CA, Medland SE, Derringer J et al: GWAS of 126,559 individuals identifies genetic variants associated with educational attainment. Science 2013; 340: 1467–1471.
Tobacco Genetics Consortium: Genome-wide meta-analyses identify multiple loci associated with smoking behavior. Nat Genet 2010; 42: 441–447.
Agrawal A, Lynskey MT, Hinrichs A et al: A genome-wide association study of DSM-IV cannabis dependence. Addict Biol 2011; 16: 514–518.
Coffee Caffeine Genetics Consortium Coffee Caffeine Genetics Consortium, Cornelis MC Coffee Caffeine Genetics Consortium, Byrne EM et al: Genome-wide meta-analysis identifies six novel loci associated with habitual coffee consumption. Mol Psychiatry 2015; 20: 647–656.
Hagenaars SP, Harris SE, Davies G et al: Shared genetic aetiology between cognitive functions and physical and mental health in UK Biobank (N=112 151) and 24 GWAS consortia. Mol Psychiatry 2016; 21: 1624–1632.
Kvale MN, Hesselson S, Hoffmann TJ et al: Genotyping informatics and quality control for 100,000 subjects in the Genetic Epidemiology Research on Adult Health and Aging (GERA) cohort. Genetics 2015; 200: 1051–1060.
Boomsma DI, Wijmenga C, Slagboom EP et al: The genome of the Netherlands: design, and project goals. Eur J Hum Genet 2014; 22: 221–227.
Scheet P, Ehli EA, Xiao X et al: Twins, tissue, and time: an assessment of SNPs and CNVs. Twin Res Hum Genet 2012; 15: 737–745.
Purcell S, Neale B, Todd-Brown K et al: PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007; 81: 559–575.
Genome of the Netherlands Consortium: Whole-genome sequence variation, population structure and demographic history of the Dutch population. Nat Genet 2014; 46: 818–825.
The 1000 Genomes Project Consortium The 1000 Genomes Project Consortium, Auton A The 1000 Genomes Project Consortium, Brooks LD et al: A global reference for human genetic variation. Nature 2015; 526: 68–74.
van Leeuwen EM, Kanterakis A, Deelen P et al: Population-specific genotype imputations using minimac or IMPUTE2. Nat Protoc 2015; 10: 1285–1296.
Zheng HF, Rong JJ, Liu M et al: Performance of genotype imputation for low frequency and rare variants from the 1000 genomes. PLoS One 2015; 10: e0116487.
Bulik-Sullivan B, Finucane HK, Anttila V et al: An atlas of genetic correlations across human diseases and traits. Nat Genet 2015; 47: 1236–1241.
Acknowledgements
This work was supported by Biobanking and Biomolecular Resources Research Infrastructure (BBMRI-NL) [184.021.007], the National Institutes of Health (NIH) [RC2MH08995], ENGAGE (NIHHEALTHF4–2007–201413), the Royal Netherlands Academy of Science Professor Award (PAH/6635) to DIB, the EMGO+ Institute for Health and Care Research, Neuroscience Campus Amsterdam, and the Avera Institute for Human Genetics. We would like to thank and acknowledge all the individuals and scientists who participated in the Genome of the Netherlands Project (GoNL). Charlie Grieser and Sahar Nohzadeh-Malakshah are employees of Affymetrix, Inc.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Ehli, E., Abdellaoui, A., Fedko, I. et al. A method to customize population-specific arrays for genome-wide association testing. Eur J Hum Genet 25, 267–270 (2017). https://doi.org/10.1038/ejhg.2016.152
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2016.152
This article is cited by
-
Integrative Multi-omics Analysis of Childhood Aggressive Behavior
Behavior Genetics (2023)
-
A comprehensive evaluation of polygenic score and genotype imputation performances of human SNP arrays in diverse populations
Scientific Reports (2022)
-
Genome-wide association study of frontotemporal dementia identifies a C9ORF72 haplotype with a median of 12-G4C2 repeats that predisposes to pathological repeat expansions
Translational Psychiatry (2021)
-
The Longitudinal Aging Study Amsterdam: cohort update 2019 and additional data collections
European Journal of Epidemiology (2020)
-
The EMIF-AD PreclinAD study: study design and baseline cohort overview
Alzheimer's Research & Therapy (2018)
