
Abstract
Fanconi anaemia (FA) is characterized by progressive bone marrow failure, congenital anomalies, and predisposition to malignancy. In a minority of cases, FA results from biallelic FANCD1/BRCA2 mutations that are associated with early-onset leukaemia and solid tumours. Here, we describe the clinical and molecular features of a remarkable family presenting with multiple primary colorectal cancers (CRCs) without detectable mutations in genes involved in the Mendelian predisposition to CRCs. We unexpectedly identified, despite the absence of clinical cardinal features of FA, a biallelic mutation of the FANCD1/BRCA2 corresponding to a frameshift alteration (c.1845_1846delCT, p.Asn615Lysfs*6) and a missense mutation (c.7802A>G, p.Tyr2601Cys). The diagnosis of FA was confirmed by the chromosomal analysis of lymphocytes. Reverse transcriptase (RT)-PCR analysis revealed that the c.7802A>G BRCA2 variation was in fact a splicing mutation that creates an aberrant splicing donor site and results partly into an aberrant transcript encoding a truncated protein (p.Tyr2601Trpfs*46). The atypical FA phenotype observed within this family was probably explained by the residual amount of BRCA2 with the point mutation c.7802A>G in the patients harbouring the biallelic FANCD1/BRCA2 mutations. Although this report is based in a single family, it suggests that CRCs may be part of the tumour spectrum associated with FANCD1/BRCA2 biallelic mutations and that the presence of such mutations should be considered in families with CRCs, even in the absence of cardinal features of FA.
Similar content being viewed by others
Introduction
Fanconi anaemia (FA) is an autosomal recessive condition characterized by congenital abnormalities, progressive pancytopenia and a predisposition to leukaemia and solid tumours.1, 2, 3 Fifteen genes, defining 15 complementation groups, have been shown to be involved in FA.4 One of them, FANCD1, corresponds to the BRCA2 gene, one of the main genes involved in autosomal dominant predisposition to breast and ovarian cancers. The proteins encoded by FA genes cooperate in the FA/BRCA pathway involved in the recognition and repair of damaged DNA.5 Disruption of FA/BRCA pathway leads to spontaneous and DNA cross-linking agents-induced chromosomal breakage.6, 7
In FA, there are typically several clinical stages related to age.8, 9 At birth and during early childhood, only physical signs are usually present with various severities, including typical but inconstant radial abnormalities. Aplastic anaemia is usually the first adverse event in patients with FA, occurring at a median age of 8–10 years. The diagnosis of FA is often made at this stage. Leukaemia occurs usually in teenagers and young adults, and solid tumours in the second decade. The more frequent solid tumours are head and neck and gynaecologic squamous cell carcinomas.10, 11 Sub-typing of FA is clinically important considering the genotype/phenotype correlations. Indeed, some groups present a more severe phenotype than others. FA subtype D1 (FA-D1), due to biallelic BRCA2 mutations and representing 3% of FA cases, is characterized by a more severe phenotype than that observed in the other groups, and FA-D1 patients often died in childhood.12 Although FA-D1 patients display less frequent bone marrow suppression and less frequent skeletal abnormalities, they present, as compared with the other FA patients an unusually high spontaneous chromosome aberration rate and early-onset malignancies including acute myeloid leukaemia (AML), Wilm’s tumour and medulloblastoma.
Here, we report a remarkable family harbouring a biallelic BRCA2 mutation and presenting with multiple malignancies including primary colorectal cancers (CRCs).
Clinical reports
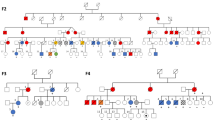
Patient 1 (III: 4) is a 33-year-old female, the first of four children born from healthy unrelated parents (Figure 1 and Supplementary Figure). No birth defect or developmental abnormalities were noticed at birth. She had an uneventful childhood. She has a healthy 12-year-old boy. She presented at 31 years of age with stomach aches progressing over 3 months with night sweating. Gastric fibroscopy with biopsies revealed an intense infiltration of CD20 and bcl-6 expressing cells, corresponding to a diffuse large B-cell lymphoma of the stomach (Ann Arbor Classification). Clinical examination and a PET scan showed cervical lymph nodes compatible with another localization of the lymphoma. The treatment of this stage III B-cell lymphoma consisted of eight courses of chemotherapy (R-CHOP: rituximab–cyclophosphamide–doxorubicin–vincristine ) including a cross-linker agent. No severe chemotherapy toxicity was noticed. Complete remission was obtained after the seventh course of chemotherapy. During the first systemic screening, scintigraphic examination showed an abdominal hyperfixation. The colonoscopic investigation revealed in the caecum, the ascending colon and sigmoid, three tumours that corresponded to three pT2N0 colorectal adenocarcinomas, which lead to colectomy. Molecular and immunochemistry analyses of the tumours showed no RER phenotype and a normal expression of the MSH2, MLH1, MSH6 and PMS2 MMR proteins. These tumours had, therefore, a microsatellite stable (MSS)phenotype. At the age of 35, four years after lymphoma and CRCs onset, a colonoscopic follow-up revealed a new rectal tumour that corresponded to a pT3N0 adenocarcinoma. The patient underwent a laparoscopic protectomy. Prophylactic hysterectomy with bilateral salpingo-oophorectomy was performed at the same time. Retrospective clinical examination revealed in the patient café-au-lait spots (CALS).

Tree of the reported family. Closed symbols indicate patients affected with FA. Open symbols indicate patients unaffected with FA. The type of cancer and age at presentation (in brackets) are given under the symbol. BRCA2 mutations are added next to the symbol. Wt: wild type.
Patient 2 (III: 5) is a female patient and the second child of the family. She also had a dizygotic twin sister (patient 3 III: 6, Figure 1). No birth defect or developmental abnormalities were noticed at birth, and she had an uneventful childhood. At 27 years of age, she presented with recurrent constipation and left iliac fossa pain. Considering the familial history, she underwent a colonoscopy that revealed in the ascending colon a tumour corresponding to a moderately differentiated pT1N0 MSS adenocarcinoma. The patient underwent a colectomy. After 2 years, the patient discovered, by self-examination in the right breast, a tumour that corresponded to a pT1bN0M0 infiltrative ductal carcinoma, positive for progesterone receptor and negative HER2 staining. The patient underwent mammectomy, followed by hormonotherapy. Retrospective clinical examination revealed CALS, mild microcephaly, enophtalmia and abnormal shape of ears. The serum AFP level was at 169 μg/l (N<10 μg/l), consistent with the levels observed in FA patients. The hemogram, including the mean cell volume, remained normal.
Patient 3 (III: 6) was the twin sister of patient 2 and died at 5 years of age from an AML. At birth, measurements were <−1 standard deviation (SD) for weight and size and <−1.5 SD for occipital frontal circumference. No congenital abnormalities had been noticed. At 5 years of age, an AML was diagnosed. Bone marrow failure or myelodysplasia were not reported before the development of AML. Before the initiation of chemotherapy, a complete blood-cell count had revealed a white blood-cell count of 45 900/mm3, haemoglobin 8.8 g/dl, mean cell volume 95 fl (within normal range for age) and the platelet count was 9 × 109/l. Blood and bone marrow were infiltrated with 77% and 85% of blastic cells, respectively. The treatment included daunorubicin (200 mg/m2/j), and aracytine (200 mg/m2/j). The first chemotherapy course was complicated by prolonged aplasia, leading to severe mucositis, disseminated fungal infection, and bacteremia, which resulted in death of the patient, 2 months after initial diagnosis. Microcephaly (<−2 SD) had been noticed in her clinical file at the time of AML diagnosis.
As indicated in the Supplementary Figure, the patients also had another 20-year-old cancer-free sister (III: 7). The father of the proband (II: 7) had a negative colonoscopic examination at 57 years of age. One of his sisters (II: 6) died from breast cancer at the age of 50 years. The mother of the proband (II: 8) had a negative mammography and a negative colonoscopy at 58 years of age. Her father (I: 3) died at 62 years of age of a liver cancer. The primary or secondary origin of this cancer was unknown.
Materials and methods
Mutational analysis of the BRCA1, BRCA2, MLH1, MSH2, MSH6, TP53 and MYH genes
Genomic DNA was extracted from peripheral blood samples, after informed consent was obtained for each patient. Point mutation screening of the BRCA1 and BRCA2 genes was performed in patient 2 (III: 5), using HRM/sequencing (BRCA2: NM_000059.3). Screening for large rearrangements of the BRCA1 and BRCA2 genes was performed using multiplex ligation-dependent probe amplification. Segregation analyses in the proband family for the mutations of BRCA2 were performed by exon 10 and 16 sequencing. Sequencing of the MLH1, MSH2, and MSH6 genes, screening for large rearrangements of MSH2 and MLH1 using multiplex ligation-dependent probe amplification, sequencing of the MLH1 promoter and determination of the MLH1 methylation status were performed in patients 1 (III: 4) and 2 (III: 5). Sequencing and screening for large rearrangements of TP53 and direct sequencing of MYH were performed in Patient 1(III: 4).
In silico prediction tools
To analyse the impact of BRCA2 c.7802A>G variant on splicing, we used the Alamut decision-support software (Interactive Biosoftware, Rouen, France) that includes the splice site prediction by neural network (NNSplice),13 splice site finder,14 Max- EntScan,15 ESE finder16 and human splicing finder17 algorithms. This software also includes three protein-function prediction tools: Align-GVGD, (http://agvgd.iarc.fr/), SIFT (http://sift.jcvi.org/), and PolyPhen-2 http://genetics.bwh.harvard.edu/pph2/.
Splicing study of the BRCA2 c.7802A>G variant
Total RNAs were isolated from the PaxGen blood RNA tube, using the RNeasy mini kit (Qiagen, Courtaboeuf, France). RNA was reverse-transcribed using random primers (Superscript (Life Technologies, Saint Aubin, France), Invitrogen (Life Technologies)) and BRCA2 cDNA was amplified, using primer F: 5′-TCACAAAGTGTGAAGAAGAACCTT-3′, primer R: 5′-AGATGCCTAAGCCCAGAAA-3′, corresponding to exons 14 and 17, respectively. RT-PCR products were electrophoresed on agarose gel, purified using the NucleoSpin Extract II kit (Macherey-Nagel, SARL, Hoerdt, France) and sequenced using the BigDye Terminator kit (Applied Biosystems, Life Technologies).
Chromosome breakage analysis
Metaphase cells from peripheral blood lymphocytes of patient 2 were G-banded after brief pretreatment in 25% pancreatin (Gibco, Life Technologies), followed by counterstaining with Wright stain. Sixty-five G-banded metaphase cells from the baseline and sixty-five metaphase cells from mitomycin (MMC) conditions were evaluated for chromosomal abnormalities, as previously described.7
Results
In this family presenting with multiple primary early-onset CRCs, analysis of MSH2, MLH1, MSH6, MYH, BRCA1 and TP53 genes revealed no germline alteration abnormality. In patient 2, early-onset breast cancers led us to analyse the BRCA genes. This analysis unexpectedly revealed two BRCA2 mutations, corresponding to a nonsense mutation c.1845_1846delCT (p.Asn615Lysfs*6) within exon 10 (Figure 2) and a missense variation c.7802A>G (p.Tyr2601Cys) within exon 16 (Figure 3). Segregation analysis in the family confirmed that each mutation was inherited from one parent. Both mutations were also found in the older sister (patient 1), who had developed two early-onset cancers. Pre-symptomatic testing revealed that the younger sister III: 5 carried the maternal BRCA2 c.1845_1846delCT mutation. In contrast to the maternal c.1845_1846delCT, which has previously been reported in the French database (http://www.umd.be/BRCA2/4DACTION/DMD_EX1/10) of the Groupe Génétique et Cancer (GGC) and is deleterious, the c.7802A>G variant, paternally inherited, was of unknown significance. It has not been reported in the GGC French mutation database, neither in the 1000 Genomes project (http://browser.1000genomes.org/) nor in dbSNP (built 137). This variation is predicted to result in a missense mutation (p.Tyr2601Cys) and in silico predictions, AGVGD (Class C55); Polyphen (score 1 probably damaging); SIFT (predict non-tolerated), classified this variant as deleterious at the protein level. Nevertheless, this genetic variation is located four nucleotides before the end of exon 16 and the different splicing prediction algorithms predicted the creation of an aberrant splicing donor site, located four nucleotides upstream of the physiological 5′ splicing site (human splicing finder scores of 74,1 versus 74,7). We therefore analysed, by RT-PCR performed on RNA extracted from patient 2 peripheral blood, the impact on splicing of the c.7802A>G variant. As indicated in Figure 4, sequencing of the RT-PCR products showed the presence of three different transcripts: Transcript A corresponded to the expected RNA expressed from the mutant BRCA2 allele carrying the c.1845_1846delCT mutation (Figure 5a). Transcripts B and C derived from the allele harbouring the substitution c.7802A>G and, as indicated by the bioinformatics prediction, transcript C was characterized by a skipping of the four last nucleotides of exon 16, owing to the creation of an aberrant splice donor site: r.7802_7805delauag. This aberrant transcript was predicted to result in a truncated protein: p.Tyr2601Trpfs*46. Comparison of the sequencing electropherogram peak heights suggested that transcripts A, B and C represent, respectively, 50%, 25% and 25% of the transcripts expressed in the blood from the patient (Figure 5b and c).

Forward and reverse BRCA2 sequence with the exon 10 mutation c.1845_1846delCT p.Asn615Lysfs*6.

Forward and reverse BRCA2 sequence with the missense variant c.7802A>G p.Tyr2601Cys in exon 16.

In silico analysis using Alamut software: appearance of a de novo 5′ splicing site (GU), located four nucleotides upstream of the physiological 5′ splicing site, suggesting a splice anomaly. The strength of this de novo 5′ splicing site is equivalent to the physiological 5′ splicing site, with scores of 74,1 versus 74,7 according to the human splicing finder.

(a) cDNA forward sequence of the mutant c.7802A>G and control patient. A: TTTTA TAGGGCTCTGTGTGACACT: allele wild type: 50% of transcript. B: TTTTG TAGGGCTCTGTGTGACACT: transcript with c.7802A>G without nucleotide deletion: r.7802a>g p.Tyr2601Cys: 25% of transcript. C: TTTTGGCTCTGTGTGACACTCCAG: transcript with c.7802A>G: truncated transcript r.7802_7805delauag p.Tyr2601Trpfs*46: 25% of transcript. (b) cDNA reverse sequence; mutant c.7802A>G. A: AGTGTCACACAGAGCCCTA TAAAATT: allele WT: 50% of transcript. B: AGTGTCACACAGAGCCCTA CAAAATT: transcript c.7802A>G without nucleotide deletion: r.7802a>g p.Tyr2601Cys: 25% of transcript. C: AGTGTCACACAGAGCCAAAATTCTTC: transcript with c.7802A>G: truncated transcript r.7802_7805delauag p.Tyr2601Trpfs*46: 25% of transcript. (c) Zoom illustrating triple peak.
In peripheral blood from patient 2, the cytogenetic analysis revealed 41% of cells with aberrations (27/65) and one triradial figure, confirming the diagnosis of FA. In the presence of a DNA cross-linker agent (MMC), an exceptionally high number of MMC-induced breaks per cell (100%) and several triradial figures were observed.
Discussion
We report in this study a family presenting a remarkable history of multiple primary cancers, including CRCs, lymphoma and breast cancer, due to biallelic BRCA2 mutations and revealing unexpectedly the diagnosis of FA. This report is the first describing the association of early-onset CRCs with FA and, therefore broadens the phenotypic spectrum associated with biallelic BRCA2 mutations. The early development of CRCs in two sisters harbouring biallelic BRCA2 mutations and the absence of detectable germline alteration of genes known to be involved in the Mendelian predisposition to CRCs are strong arguments establishing a possible link between the development of early-onset CRCs and biallelic BRCA2 mutations. Remarkably, in this family, the diagnosis of early-onset breast cancer in patient 2 led us to analyse the BRCA genes that revealed the presence of a biallelic alteration of BRCA2, whereas there was no obvious clinical feature suggestive of FA. Indeed, patient 1 and 2 did not display any haematologic abnormality at 29 and 33 years, and did not have any radial defects. Only careful retrospective examination subsequently revealed mild microcephaly, enophtalmia, and CALS. FA diagnosis was firmly confirmed in patient 2 who displayed highly increased chromosomal breakage, as well as constitutionally increased alpha-foetoprotein. In patient 3, the diagnosis of FA was very likely considering: i) the development of an AML, ii) the chemotherapic toxicity with persistent aplasia after first induction, iii) the low platelet count, and iv) the occipito-frontal circumference at −2 SD. Thus, the current data were notified to the french database of the ‘GGC’ (http://www.umd.be/BRCA2/) and as the combination of two BRCA2 mutations reported here results in FA, the BRCA2 variant c.7802A>G will be classified as deleterious.
In FA, all the genotype/phenotype correlation studies indicate that the FA-D1 group, like the FA-N group, can be distinguished from the other groups by a more severe phenotype and a high mortality rate in early childhood.12, 18 Table 1 gives a review of the 30 patients from 24 families with biallelic BRCA2 mutations and whose phenotype had previously been described in details family.12, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28 Among these patients, the females and males were equally affected and the mean age of FA diagnosis was 1.9 years (range, 0.1–5.2 years). Most of the patients were diagnosed as FA because of their typical physical features (68%). Ten patients had severe congenital malformations (cardiac, brain or intestinal abnormalities), including five VATER associations (Vertebral defects, Anal atresia, Tracheoesophageal fistula with esophageal atresia, Renal, and Radial dysplasia). Myelodysplasia or bone marrow failure was reported in eight patients. Among the patients, 19 (63.3%) were dead at a mean age of 3.9 years (range 1.4–12 years). Among the 11 (36.6%) alive at the time of report, the mean age was of 5.84 years (range 0.8–30 years). Fourteen died because of the progression of malignancy and five died because of a severe toxicity of chemotherapy. Malignancies included brain tumours (17 cases corresponding mainly to medulloblastoma), AML (13 cases), Wilm’s tumours (eight cases), and acute lymphoblastic leukaemia (four cases). At 2 years of age, half of the patients (15/30) had already developed a cancer. At 5 years of age, all but one had developed at least one tumour, and in 11 cases (34%) multiple cancers occurred. In agreement with these data, Alter et al12 estimated that, in FA patients, the cumulative risk of malignancy, at 5.2 years of age, is 97%. BRCA2 is directly involved in DNA double-strand break DNA repair, by homologous recombination, which might explain the severity of the phenotype in FA-D1 group.5 It has been first speculated that individuals with BRCA2 biallelic mutations will only be viable if they carried at least one ‘hypomorphic’ allele, such as a missense mutation or a truncating mutation close to the 5′ terminus,21 but this was subsequently disproved.28 The spectrum of mutations in FA-D1 patients is not consistent with the mutation spectrum observed in breast cancer families related to BRCA2 mutations, suggesting that some combinations of deleterious mutations are not viable. Furthermore, and despite the high population prevalence of BRCA2 heterozygous carriers of the c.6147delT mutation in the Ashkenazim, no homozygous carrier has been reported.29 Table 1 shows only eight cases carrying one truncating mutation and one missense mutation. More than half of the patients had two truncating mutations (18/30; 60%). HSC62 was the only patient who reached adulthood without a history of cancer (Table 1), and he was homozygous for an in-frame deletion of four codons that might explain this mild phenotype.21
Genetic variations, which cause aberrant splicing, may represent up to 50% of all the deleterious mutations.30 Depending on tissue localization and/or stage of development, pre-mRNA transcripts may be differentially spliced. We showed that the BRCA2 c.7802A>G variant detected in this study produced 50% of transcripts harbouring a premature stop codon (r.7802_7805delauag) and 50% of transcripts with a p.Tyr2601Cys mutation. In patients 1 and 2 harbouring both BRCA2 mutations and exhibiting an atypical FA phenotype mostly characterized by early-onset tumours, at least 75% of the BRCA2 transcripts are predicted to code for a truncated protein and 25% for the p.Tyr2601Cys variant. The 2601 residue is located within the helical domain (located from 2478 to 2668 amino acid) of BRCA2 in the C-terminal domain. The absence of classical FA features in patients 1 and 2 suggests that this variant protein either partially conserved the BRCA2 function or had a normal function and that its amount is sufficient to prevent the appearance of bone marrow failure in early childhood and classical FA features. The development in patient 3 of early-onset AML, corresponding to a more severe phenotype, might be explained by a phenotypic variability of the mutation, which is commonly observed with splicing alterations. Despite identical BRCA2 mutations, divergent phenotype and clinical courses within these siblings might be also explained by the differential genetic background, influences of modifier genes, environmental effects or chance effects.
The results of the cDNA sequencing suggest that the aberrant mRNAs A and C (RT-PCR on blood sample Paxgene RNA) are stable in our family (Figure 5). The explanation for this finding can be as follows: an exon–exon junction complex of proteins deposited approximately 20–24 nt upstream of exon–exon junctions during pre-mRNA splicing is considered to be a primary determinant of nonsense-mediated decay (NMD) in mammalian cells; and as a rule, NMD is generally triggered when the translation termination at a nonsense codon is located more than approximately 50–55 nt upstream of an exon–exon junction.31 In our patient, the aberrant mRNAs A and C are both containing a premature translation termination codon (PTC) located in the area of approximately 50–55 nt upstream of an exon–exon junction and thus are not supposed to trigger NMD. For the mutant c.1845_1846delCT p.Asn615Lysfs*6, the PTC is located 55 nt before the end of the exon 10 (exon 10–exon 11 junction), and for the mutant c.7802A>G p.Tyr2601Cys, the aberrant transcript r.7802_7805delauag p.Tyr2601Trpfs*46 introduces the PTC approximately 35 nt before the end of the exon 17 (exon 17–exon 18 junction).
Finally, the development of early-onset CRCs in two FA patients might be indicative of an interaction between BRCA2 and MMR proteins. Heterozygous mutations of the MMR genes constitute the genetic basis of the Lynch syndrome, the main cause of the Mendelian form of CRCs, and the development of tumours in Lynch syndrome results from the RER phenotype leading to somatic mutations within target genes containing repeats.32, 33 Remarkably, homozygous inactivation of the MMR genes, corresponding to the mismatch-repair deficiency syndrome (CMMR-D), was subsequently identified in children or young adults presenting with early-onset brain tumours, leukaemia or lymphoma, malignancies of the digestive tract and CALS, and this phenotype spectrum was a first clinical indication that the MMR genes were also involved in DNA double-strand homologous recombination, an hypothesis that was later confirmed.29 Several studies have already established an interaction between MMR and BRCA2 proteins. The FANCJ protein has been shown to interact with MLH1 and disruption of this interaction resulted in hypersensitivity to inter-strand cross-linking agents.34 The pivotal FA protein FANCD2 was subsequently shown to interact with MSH2 and MLH1.35 Our report indicates that the presence of biallelic BRCA2 mutations should be considered in families with early-onset breast and CRCs, even in the absence of cardinal features of FA.
References
Joenje H, Patel KJ : The emerging genetic and molecular basis of Fanconi anaemia. Nat Rev Genet 2001; 2: 446–457.
Kutler DI, Singh B, Satagopan J et al: A 20-year perspective on the International Fanconi Anaemia Registry (IFAR). Blood 2003; 101: 1249–1256.
Tischkowitz MD, Hodgson SV : Fanconi anaemia. J Med Genet 2003; 40: 1–10.
de Winter JP, Joenje H : The genetic and molecular basis of Fanconi anaemia. Mutat Res 2009; 668: 11–19.
Levitus M, Joenje H, de Winter JP : The Fanconi anaemia pathway of genomic maintenance. Cell Oncol 2006; 28: 3–29.
Schroeder TM, Anschutz F, Knopp A : [Spontaneous chromosome aberrations in familial panmyelopathy]. Humangenetik 1964; 1: 194–196.
Sasaki MS, Tonomura A : A high susceptibility of Fanconi's anaemia to chromosome breakage by DNA cross-linking agents. Cancer Res 1973; 33: 1829–1836.
Rosenberg PS, Greene MH, Alter BP : Cancer incidence in persons with Fanconi anaemia. Blood 2003; 101: 822–826.
Shimamura A, Alter BP : Pathophysiology and management of inherited bone marrow failure syndromes. Blood Rev 2010; 24: 101–122.
Alter BP : Cancer in Fanconi anaemia, 1927-2001. Cancer 2003; 97: 425–440.
Rosenberg PS, Huang Y, Alter BP : Individualized risks of first adverse events in patients with Fanconi anaemia. Blood 2004; 104: 350–355.
Alter BP, Rosenberg PS, Brody LC : Clinical and molecular features associated with biallelic mutations in FANCD1/BRCA2. J Med Genet 2007; 44: 1–9.
Reese MG, Eeckman FH, Kulp D, Haussler D : Improved splice site detection in Genie. J Comput Biol 1997; 4: 311–323.
Shapiro MB, Senapathy P : RNA splice junctions of different classes of eukaryotes: sequence statistics and functional implications in gene expression. Nucleic Acids Res 1987; 15: 7155–7174.
Yeo G, Burge CB : Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol 2004; 11: 377–394.
Cartegni L, Wang J, Zhu Z, Zhang MQ, Krainer AR : ESEfinder: A web resource to identify exonic splicing enhancers. Nucleic Acids Res 2003; 31: 3568–3571.
Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, Beroud C : Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res 2009; 37: e67.
Neveling K, Endt D, Hoehn H, Schindler D : Genotype-phenotype correlations in Fanconi anaemia. Mutat Res 2009; 668: 73–91.
Faivre L, Portnoi MF, Pals G et al: Should chromosome breakage studies be performed in patients with VACTERL association? Am J Med Genet A 2005; 137: 55–58.
Hirsch B, Shimamura A, Moreau L et al: Association of biallelic BRCA2/FANCD1 mutations with spontaneous chromosomal instability and solid tumors of childhood. Blood 2004; 103: 2554–2559.
Howlett NG, Taniguchi T, Olson S et al: Biallelic inactivation of BRCA2 in Fanconi anaemia. Science 2002; 297: 606–609.
Wagner JE, Tolar J, Levran O et al: Germline mutations in BRCA2: shared genetic susceptibility to breast cancer, early onset leukaemia, and Fanconi anaemia. Blood 2004; 103: 3226–3229.
Dewire MD, Ellison DW, Patay Z, McKinnon PJ, Sanders RP, Gajjar A : Fanconi anaemia and biallelic BRCA2 mutation diagnosed in a young child with an embryonal CNS tumour. Pediatr Blood Cancer 2009; 53: 1140–1142.
Ikeda H, Matsushita M, Waisfisz Q et al: Genetic reversion in an acute myelogenous leukaemia cell line from a Fanconi anaemia patient with biallelic mutations in BRCA2. Cancer Res 2003; 63: 2688–2694.
Meyer S, Fergusson WD, Oostra AB et al: A cross-linker-sensitive myeloid leukaemia cell line from a 2-year-old boy with severe Fanconi anaemia and biallelic FANCD1/BRCA2 mutations. Genes Chromosomes Cancer 2005; 42: 404–415.
Myers K, Davies SM, Harris RE et al: The clinical phenotype of children with Fanconi anaemia caused by biallelic FANCD1/BRCA2 mutations. Pediatr Blood Cancer 2012; 58: 462–465.
Offit K, Levran O, Mullaney B et al: Shared genetic susceptibility to breast cancer, brain tumors, and Fanconi anaemia. J Natl Cancer Inst 2003; 95: 1548–1551.
Reid S, Renwick A, Seal S et al: Biallelic BRCA2 mutations are associated with multiple malignancies in childhood including familial Wilms tumour. J Med Genet 2005; 42: 147–151.
Rahman N, Scott RH : Cancer genes associated with phenotypes in monoallelic and biallelic mutation carriers: new lessons from old players. Hum Mol Genet 2007; 16 Spec No 1: R60–R66.
Cartegni L, Chew SL, Krainer AR : Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet 2002; 3: 285–298.
Matsuda D, Sato H, Maquat LE : Chapter 9. Studying nonsense-mediated mRNA decay in mammalian cells. Methods Enzymol 2008; 449: 177–201.
Grady WM, Markowitz SD : Genetic and epigenetic alterations in colon cancer. Annu Rev Genomics Hum Genet 2002; 3: 101–128.
Muller A, Fishel R : Mismatch repair and the hereditary non-polyposis colorectal cancer syndrome (HNPCC). Cancer Invest 2002; 20: 102–109.
Peng M, Litman R, Xie J, Sharma S, Brosh RM Jr., Cantor SB : The FANCJ/MutLalpha interaction is required for correction of the cross-link response in FA-J cells. EMBO J 2007; 26: 3238–3249.
Williams SA, Wilson JB, Clark AP et al: Functional and physical interaction between the mismatch repair and FA-BRCA pathways. Hum Mol Genet 2011; 20: 4395–4410.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Author contributions
M-MP, SG, FC, CL, CH, EF, AD, GC, SEC, PF, LF, AB, CL, J-LJ provided clinical data. ED-C, JS, MB, CC, CC, LM, SL, LA, PC, FM, TF, PJ and LF contributed to conception and design, acquisition of data, and analysis and interpretation of data. ED-C, JS, TF, PJ, LF contributed to writing the manuscript.
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Degrolard-Courcet, E., Sokolowska, J., Padeano, MM. et al. Development of primary early-onset colorectal cancers due to biallelic mutations of the FANCD1/BRCA2 gene. Eur J Hum Genet 22, 979–987 (2014). https://doi.org/10.1038/ejhg.2013.278
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2013.278
Keywords
This article is cited by
-
Clinical consequences of BRCA2 hypomorphism
npj Breast Cancer (2021)

{kind=link}