
Abstract
Autosomal recessive Adams–Oliver syndrome was diagnosed in three remotely related Bedouin consanguineous families. Genome-wide linkage analysis ruled out association with known Adams–Oliver syndrome genes, identifying a single-homozygosity ∼1.8-Mb novel locus common to affected individuals (LOD score 3.37). Whole-exome sequencing followed by Sanger sequencing identified only a single mutation within this locus, shared by all affected individuals and found in patients from five additional apparently unrelated Bedouin families: a 1-bp deletion mutation in a predicted alternative splice variant of EOGT, leading to a putative truncated protein. RT-PCR demonstrated that the EOGT-predicted alternative splice variant is ubiquitously expressed. EOGT encodes EGF-domain-specific O-linked N-acetylglucosamine transferase, responsible for extracellular O-GlcNAcylation of epidermal growth factor-like domain-containing proteins, and is essential for epithelial cell-matrix interactions. F-actin staining in diseased fibroblasts showed apparently intact cell cytoskeleton and morphology, suggesting the EOGT mutation acts not through perturbation of cytoskeleton but through other mechanisms yet to be elucidated.
Similar content being viewed by others

Introduction
Adams–Oliver syndrome (AOS; MIM 100300) is a multiple congenital anomaly syndrome characterized by vertex scalp defects (aplasia cutis congenita; ACC), with terminal transverse limb defects (TTLD).1 Limb defects range from nail dystrophy and syndactyly to asymmetric shortening of hands and feet, to absence of digital extremities.2 Deformities of the skull are found in some cases. Additional clinical manifestations can include congenital cardiac malformations, vascular defects, and neurological abnormalities.2 AOS has been described in both autosomal dominant and recessive modes of inheritance.2 Recent identification of AOS-causing dominant mutations in ARHGAP31 (MIM 610911)3 and recessive mutations in DOCK6 (MIM 614194)4 confirms both modes of inheritance and highlights Cdc42/Rac1 regulation of actin cytoskeleton during early human development. AOS can be caused also by dominant mutations in RBPJ (MIM 147183), a transcriptional regulator of the NOTCH signaling pathway, further demonstrating the genetic heterogeneity of the phenotype.5 As some features of AOS are consistent with vascular disruption pathogenesis,2 it is yet unclear how disrupted Cdc42/Rac1 or NOTCH signaling intersects in the pathogenesis of AOS. Identification of additional genes implicated in AOS might elucidate common molecular pathways leading to this disease.
Materials and methods
Patients
We studied eight affected individuals of three remotely related consanguineous families of the same Bedouin tribe (Figure 1a) and seven individuals of five additional Bedouin families of other tribes, originated from the same clan (Figure 1b). Following Soroka Medical Center IRB approval and informed consent, DNA samples were obtained from all available affected family members, their siblings, and obligatory carrier parents.

Clinical presentation of AOS. (a) Pedigrees (P1, P2 and P3) of three affected kindred of the same Bedouin tribe, showing segregation of the c.1074delA mutation. For tested individuals, the c.1074delA mutation status of each allele is marked as wild-type (WT) or mutant (M). (b) Pedigrees of five apparently unrelated affected kindred of other tribes of the same large clan (P4, P5, P6, P7, P8), showing segregation of the c.1074delA mutation. (c-f) Characteristic phenotype of AOS. (c) Extremely severe ACC with dilated scalp veins and underlying bone defect, resulting in uncontrolled hemorrhage and death (P2-IV:8). (d) Bilateral incomplete syndactyly in toes 2–3–4 (P1-IV:4). (e) Severe ACC with dilated scalp veins and underlying bone defect. (f) Although most skull defects close spontaneously over a period of months, some remain partially open. Three dimensional CT scan showing long-term residual bone defect (P2-IV:6).
Linkage analysis
Genome-wide linkage analysis was done using Affymetrix GeneChip Human Mapping 250K Array Nsp (Affymetrix, Santa Clara, CA, USA) as previously described,6 testing one affected individual of each family (P1-IV:6, P2-IV:6, and P3-IV:1). Genotype data were analyzed using HomozygosityMapper (http://www.homozygositymapper.org/), and multipoint LOD score was calculated as previously described,6 using SUPERLINK (http://cbl-hap.cs.technion.ac.il/superlink-snp/). Four informative markers were used for the analysis: chr3:68907579–68907912, D3S1296, D3S1566, and D3S2454.
Whole-exome sequencing
Whole-exome sequencing of individual IV:6 of pedigree P1 (Figure 1a) was performed using HiSeq2000 (Illumina, San Diego, CA, USA) and analyzed as previously described.6 All uncovered regions in the disease-associated interval were PCR amplified and Sanger sequenced using the ABI PRISM 3730 DNA Analyser (Applied Biosystems, Foster city, CA, USA).
Mutation detection—restriction analysis
MnlI restriction analysis gave differential cleavage products for the EOGT wild-type and mutant sequences: 223 bp in the wild-type versus 188 bp and 34 bp fragments in the mutant. PCR amplification primers were as follows: forward 5′-AAGCAATGCAAAATGGAGATT-3′; reverse 5′-CACAAAAACATGGTCTCCTGA-3′.
RT-PCR
EOGT expression was determined using PCR amplification of cDNA from various human tissues and cells. The primer sets used flank exons common to both the 15 and the 18 exon splice variants of EOGT: forward 5′-CAGAGAGAGGCTGGAGGAGA-3′; reverse 5′-TTTTCAGTGCATTTACAAGCTCA-3′.
F-Actin staining
Fibroblasts were stained for F-actin using phalloidin (Alexa Fluor 546; Invitrogen Corporation, Carlsbad, CA, USA). Cells were stained and visualized using confocal microscopy as previously described.7
Results
All studied patients were diagnosed with autosomal recessive AOS. Apart from the aplasia cutis, the phenotype of all patients was very minimal (Table 1 and Figure 1c–f): all had scalp skin defects to different extents and limb defects ranging from minimal nail deformities to syndactyly mainly of toes. Some had skull defects at the vertex and dilated scalp veins. No further anomalies were found.
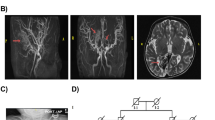
Genome-wide linkage analysis of the related families P1, P2, and P3 (Figure 1a) followed by homozygosity mapping identified a single 1.8-Mb novel locus between SNPs rs1602197 and rs4974301 on chromosome 3p14.1-13 (Chr3:68 831 371–70 639 463; GRCh37/hg19) that was common to all tested patients (Figure 2a). Genotyping of the three pedigrees combined (Figure 1a) yielded a maximum LOD score of 3.37. All affected individuals shared a common homozygous haplotype between markers chr3:68907579-68907912 and D3S2454, supporting a common ancestral origin for this locus. Whole-exome sequencing of an affected individual fully covered the 1.8-Mb interval, except for segments of EOGT (MIM 614789). Sanger sequencing of those segments identified a single homozygous mutation (Figure 2b): c.1074delA (p. Gly359Aspfs*28) in exon 13 of a predicted alternative splice variant of EOGT (RefSeq NM_173654.1; ENST00000383701.3), leading to a frameshift and premature stop codon. This predicted alternative splice variant is composed of 18 exons, similar to characterized EOGT orthologs in the mouse and drosophila.8 The mutation was not found in any SNP or mutation database. Restriction analysis using MnlI demonstrated that the mutation was common to all affected individuals in the three pedigrees, and that all obligatory carriers in those pedigrees were indeed heterozygous for the mutation (Figure 1a). None of the 100 nonrelated Israeli Bedouins controls of other clans carried the mutation. Screening of additional AOS families with no known relation to the cohort studied yet of the same clan (Figure 1b) identified five additional unrelated consanguineous Bedouin AOS families with the EOGT c.1074delA mutation.

Molecular studies of the affected kindred. (a) HomozygosityMapper genome-wide linkage analysis of the three families revealed only a single region shared by all affected individuals tested (P1-IV:6, P2-IV:6 and P3-IV:1), on chromosome 3 (red). (b) Sequence chromatogram of the c.1074delA mutation with a control tracing for comparison. Red arrow indicates the position of the mutation. Note that the NCBI database contains only alternative splice variant RefSeq NM_173654.1, according to which the nomenclature of the mutation would be c.832-791delA (instead of c.1074delA). (c) RT-PCR demonstrating ubiquitous expression of EOGT in human tissues and cells. Expected RT-PCR product sizes are 548 bp for the RefSeq 15-exon splice variant (RefSeq NM_173654.1) or 800 bp for 18-exon splice variant (ENST00000383701.3). (d) Patients and control fibroblast cells visualized by immunofluorescence microscopy for Phalloidin staining of F-actin (red) and DAPI (blue) stained nuclei. (i, ii) Control fibroblasts showing typical spindle appearance. (iii) Close-up view of control fibroblast. (iv, v) Patient fibroblasts showing appearance similar to control fibroblasts. (vi) Close-up view of patient’s fibroblast.
PCR amplification of cDNA, from various human tissues and cells, showed a single product that was ubiquitously expressed (Figure 2c) and represents the 18-exon splice variant (ENST00000383701.3), which has no representation in RefSeq. F-actin staining in diseased fibroblasts showed typical normal appearance and well-organized actin cytoskeleton filaments, similar to normal control fibroblasts, with no unusual rounded cell shapes or the perturbed actin cytoskeleton and lamellipodia (Figure 2d).
Discussion
This study, together with a parallel independent study by Shaheen et al.,9 which identified three EOGT mutations in different Arab families, defines EOGT as a novel Adams–Oliver syndrome gene. Both studies identified a common c.1074delA founder mutation in the Arab world. It should be noted that the ancestors of many of the Bedouins of southern Israel migrated from the Arabian Peninsula before the spread of the Islam around 700 AC, forming several large clans (including the clan in this study) and carry similar founder mutations.6 The c.1074delA mutation was not found in 100 controls of Bedouins of various tribes of non-related clans from southern Israel, suggesting that it is present in only a specific cohort within that community.
The ubiquitous expression of the 18-exon splice variant (ENST00000383701.3) in various human tissues (Figure 2c) provides supporting evidence that the c.1074delA mutation is expected to have a significant effect on the encoded protein. In line with the Adams–Oliver syndrome phenotype, Eogt mRNA is highly expressed in the developing limb buds of mice, showing a digit condensation pattern in the limbs by E12.5.9 EOGT encodes EGF-domain-specific O-linked N-acetylglucosamine (O-GlcNAc) transferase, which is responsible for extracellular O-GlcNAcylation of epidermal growth factor-like domain-containing proteins.8 O-GlcNAc modification is a unique dynamic post-transcriptional modification that regulates a variety of cellular processes including intracellular signaling, transcription, and protein stability.10 Drosophila Eogt mutants display defects in the apical extracellular matrix (aECM), including separation of the epidermis from the chitin layers and wing blistering.10 Among Eogt targets are Notch and Dumpy, a membrane-anchored extracellular protein required for epithelial cell-matrix interaction. Eogt drosophila mutants display apparently intact Notch signaling, despite Notch not being O-GlcNAc modified. However, Eogt is essential for Dumpy-dependent epithelial cell–matrix interactions.10 This suggests that dysfunction of EOGT in humans might cause AOS by effecting cell–cell or cell–matrix interactions, processes that are perturbed by actin cytoskeletal defects as previously described in AOS affected individuals with ARHGAP31 and DOCK6 mutations.3, 4 It is noteworthy that F-actin staining in our EOGT mutant fibroblasts showed apparently intact cell cytoskeleton and morphology. There is growing evidence that the AOS phenotype might be mediated through the NOTCH signaling pathway: AOS-causing RBPJ mutations are assumed to effect NOTCH signaling,5 and drosophila Notch ligands Delta and Serrate, as well as mammalian Notch1 are O-GlcNAc-modified by Eogt.8, 11 Moreover, heterozygosity for either Notch or for several members of the canonical Notch signaling pathway suppresses wing blistering formation caused by Eogt knockdown in drosophila.11 It should be noted, however, that Notch signaling in drosophila Eogt mutants is apparently intact, with Eogt mutants showing no neurogenic phenotypes characteristic of Notch mutants.10, 11 One should keep in mind, though, that the case might be different in mammals and further studies in this regard are warranted.
Recent evidence that the EOGT motif is shared by many proteins, including several that reside in the extracellular matrix,12 suggests that there are yet further possibilities as to the molecular pathways through which perturbed EOGT function might lead to AOS. Future studies of EOGT targets, which show an overlapping expression pattern during mammalian development, might open new venues in our understanding of this rare congenital developmental anomaly.
References
Adams FH, Oliver CP : Hereditary deformities in man due to arrested development. J Hered 1945; 36: 3–7.
Snape KM, Ruddy D, Zenker M et al: The spectra of clinical phenotypes in aplasia cutis congenita and terminal transverse limb defects. Am J Med Genet A 2009; 149A: 1860–1881.
Southgate L, Machado RD, Snape KM et al: Gain-of-function mutations of ARHGAP31, a Cdc42/Rac1 GTPase regulator, cause syndromic cutis aplasia and limb anomalies. Am J Hum Genet 2009; 88: 574–585.
Shaheen R, Faqeih E, Sunker A et al: Recessive mutations in DOCK6, encoding the guanidine nucleotide exchange factor DOCK6, lead to abnormal actin cytoskeleton organization and Adams-Oliver syndrome. Am J Hum Genet 2011; 89: 328–333.
Hassed SJ, Wiley GB, Wang S et al: RBPJ mutations identified in two families affected by Adams-Oliver syndrome. Am J Hum Genet 2012; 91: 391–395.
Volodarsky M, Markus B, Cohen I et al: A Deletion Mutation in TMEM38B Associated with Autosomal Recessive Osteogenesis Imperfecta. Hum Mutat 2013; 34: 582–586.
Cohen I, Birnbaum RY, Leibson K, Taube R, Sivan S, Birk OS : ZNF750 is expressed in differentiated keratinocytes and regulates epidermal late differentiation genes. PLoS One 2012; 7: e42628.
Sakaidani Y, Ichiyanagi N, Saito C et al: O-linked-N-acetylglucosamine modification of mammalian Notch receptors by an atypical O-GlcNAc transferase Eogt1. Biochem Biophys Res Commun 2012; 419: 14–19.
Shaheen R, Aglan M, Keppler-Noreuil K et al: Mutations in EOGT confirm the genetic heterogeneity of autosomal-recessive Adams-Oliver Syndrome. Am J Hum Genet 2013; 92: 598–604.
Sakaidani Y, Nomura T, Matsuura A et al: O-linked-N-acetylglucosamine on extracellular protein domains mediates epithelial cell-matrix interactions. Nat Commun 2011; 2: 583.
Muller R, Jenny A, Stanley P : The EGF repeat-specific O-GlcNAc-transferase Eogt interacts with Notch signaling and pyrimidine metabolism pathways in Drosophila. PLoS One 2013; 8: e62835.
Alfaro JF, Gong CX, Monroe ME et al: Tandem mass spectrometry identifies many mouse brain O-GlcNAcylated proteins including EGF domain-specific O-GlcNAc transferase targets. Proc Natl Acad Sci USA 2012; 109: 7280–7285.
Acknowledgements
The study was supported by the Israel Science Foundation (ISF grant 1689/12) and through the Kahn Family Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Cohen, I., Silberstein, E., Perez, Y. et al. Autosomal recessive Adams–Oliver syndrome caused by homozygous mutation in EOGT, encoding an EGF domain-specific O-GlcNAc transferase. Eur J Hum Genet 22, 374–378 (2014). https://doi.org/10.1038/ejhg.2013.159
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2013.159
Keywords
This article is cited by
-
Genetics of Aplasia Cutis Reveal Novel Regulators of Skin Morphogenesis
Journal of Investigative Dermatology (2015)
