
Abstract
The steroidogenic factor 1 (SF1) protein, encoded by the NR5A1 gene, plays a central role in gonadal development and steroidogenesis. Mutations in NR5A1 were first described in patients with primary adrenal insufficiency and 46,XY disorders of sexual development and later also in men with hypospadias, bilateral anorchia and micropenis and women with primary ovarian insufficiency. Recently, heterozygous missense mutations were found in 4% of infertile men with unexplained reduced sperm counts living in France, but all mutation carriers were of non-Caucasian ancestry. Therefore, we performed a comprehensive NR5A1 sequence analysis in 488 well-characterised predominantly Caucasian patients with azoo- or severe oligozoospermia. Two-hundred-thirty-seven men with normal semen parameters were sequenced as controls. In addition to several synonymous variants of unclear pathogenicity, three heterozygous missense mutations predicted to be damaging to SF1 protein function were identified. The andrological phenotype in infertile but otherwise healthy mutation carriers seems variable. In conclusion, mutations altering SF1 protein function and causing spermatogenic failure are also found in men of German origin, but the prevalence seems markedly lower than in other populations.
Similar content being viewed by others


Introduction
About 10–15% of couples have problems conceiving and in half of cases a male (co-)factor can be found upon clinical workup. Male infertility is mostly caused by spermatogenetic failure, clinically noted as oligo- or azoospermia, and a substantial genetic contribution can be assumed from familial studies and from animal—mostly mouse knockout—models.1 Overall, a large percentage of human male infertility is estimated to be caused by mutations in genes involved in germ cell production and function.4 However, with the current routine workup of the infertile male comprising karyotype analysis and Y-chromosomal AZF deletion screening,5 a genetic cause can only be elucidated in about 5% of unselected and 20% of azoospermic patients.6, 7, 8
Although many efforts have been undertaken to identify new causative genes for human male infertility, no candidate gene has been recognised to be administrable in patient care so far.9, 10, 11 In contrast, Bashamboo et al.12 found mutations in the NR5A1 gene (MIM 184757) encoding steroidogenic factor 1 (SF1) protein in a substantial fraction of 4% of infertile men (N=315) with unexplained reduced sperm counts and sperm concentrations below 1 million/ml. All of these were missense mutations leading to reduced protein function in vitro.12
The SF1 protein has a central role in gonadal development and steroidogenesis, and both male and female Nr5a1 null mice exhibit adrenal agenesis, have female internal genitalia, and gonadal agenesis.13 Mutations in NR5A1 were first described in patients with primary adrenal insufficiency and 46,XY disorders of sexual development and recently also in women with primary ovarian insufficiency and boys with hypospadias, bilateral anorchia and/or micropenis broadening the phenotypic spectrum.14
As all mutations in otherwise healthy infertile men were found in subjects of non-Caucasian ancestry and the spermatogenic phenotype in carriers remains elusive, we performed comprehensive NR5A1 mutation screening in a large group of well-characterised infertile men predominantly of German origin.
Materials and Methods
Study population
Four-hundred-eighty-eight patients who attended the Department of Clinical Andrology, Centre of Reproductive Medicine and Andrology, University Clinic Münster, a tertiary-referral centre for infertility were retrospectively selected using the Androbase database.15 Only patients with severe oligozoospermia (sperm concentration <5 million/ml, N=218) or non-obstructive azoospermia (N=270) were included. Definite causes for male infertility (karyotype anomalies, Y-chromosomal AZF deletions, oncologic disease and/or chemo-/radiotherapy) were excluded. To explore an association of NR5A1 mutations with disturbances of testicular decent, patients with a history of cryptorchidism were part of the study group (N=110 of the oligozoospermic and N=76 of the azoospermic men). As controls, 237 men without cryptorchidism and with normozoospermia (≥39 million total sperm, ≥32% progressive motility and ≥4% normal morphology) according to the current World Health Organisation reference ranges16 were selected. Men were predominantly of Caucasian ethnicity and German origin (90%) according to self report. Ejaculates were obtained by masturbation and all semen parameters were determined in accordance with WHO guidelines.16 Semen analyses were under constant internal and external quality control.
Ethics statement
All participants gave informed written consent for evaluation of their clinical data and genetic analysis of their donated DNA samples, according to protocols approved by the Ethics Committee of the Medical Faculty in Münster.
Mutation analysis
Genomic DNA was extracted from EDTA-preserved blood using the standard techniques. All DNA probes were whole-genome amplified by using the Illustra GenomiPhi V2 DNA Amplification Kit (GE Healthcare Life Sciences, Chalfont St Giles, UK), according to the manufacturer’s description. PCR was carried out in a volume of 20 μl with ∼200 ng DNA and specific primers for all exons of NR5A1 (exons 1–7; NM_004959.4) as described in Supplementary Table 1. Exon 4 was divided into two overlapping PCR formats. For sequencing, the PCR products were treated with ExoSAP-IT (USB Corporation, Cleveland, OH, USA). The sequencing reaction was carried out using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Carlsbad, CA, USA) and analysed on a 3730 DNA Analyser (Applied Biosystems). Mutations were confirmed by sequencing the corresponding exon again in both directions with the original DNA sample of the patient. PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/) and two splice site prediction programmes (GeneSplicer, http://www.cbcb.umd.edu/software/GeneSplicer/gene_spl.shtml and Human Splicing Finder, http://www.umd.be/HSF/) were used for in silico analyses.
Results
In total, ten different NR5A1 mutations were identified in 26 of the 488 patients and in seven of the 237 controls (Table 1). Five of these mutations were novel and the others were previously described and present in dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP/) with very low minor allele frequencies except one common polymorphism.
The previously described missense mutation c.437G>C (P.Gly146Ala, rs1110061) was present in 16 patients (14 hetero-, 2 homozygous) and 5 controls (four hetero-, one homozygous). Neither allele frequencies (χ2-test P=0.41) nor genotype distribution (P=0.62) differed significantly between patients and controls. Moreover, this polymorphism was exclusively found in patients without a history of cryptorchidism (Table 2).
Synonymous mutations were analysed for possible effects on splicing, but the in silico predictions did not hint to strong effects for any one of the variants. These mutations were, however, more prevalent in patients than controls (1.0 vs 0.4%).
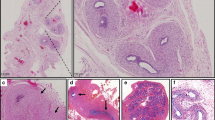
The remaining three private missense mutations were found in one infertile man each. All of these lead to substitutions of highly conserved amino acids and are predicted to be damaging to SF1 protein function (Figure 1). Clinical details of the carriers are described in Table 3 and the testicular histology of two of the patients is presented in Figure 2. No testicular biopsy was available for the patient carrying the mutation p.Ile323Thr.

Alignment of amino-acid sequences of NR5A1 for the three missense mutations showing high conservation.

Representative testicular histology of patients with NR5A1 missense mutations. (a) Case 1 carrying p.Gly165Arg showing meiotic arrest with spermatocytes as the most advanced germ cells and (b) case 2 carrying P.Asp257Asn exhibiting mixed atrophy with predominantly Sertoli-cell-only tubules.
The overall prevalence of NR5A1 mutations not found in controls was 1.6% (8/488) in our patient group and comparable between men with severe oligozoospermia (4/218, 1.8%) and azoospermia (4/270, 1.5%, Table 2). Missense mutations (excluding c.437G>C) were exclusively found in patients with a frequency of 0.6% (3/488). The highest sperm concentration found in a mutation carrier was 0.3 million/ml and if only patients with sperm concentrations below 1 million/ml were taken into account (66 patients had sperm concentrations between 1 and 5 million/ml), probably pathogenic NR5A1 mutations were found in 0.7% (3/422). None of these percentages changed significantly when only Caucasian patients were analysed.
Discussion
Missense mutations in NR5A1 have recently been described as a novel cause for male infertility.12 In our comprehensive mutation screen of 488 well-characterised patients, we identified only three missense mutations likely causative for the phenotypes of azoo-, crypto- and severe oligozoospermia according to the in silico analyses. Compared with the mutations found in the study by Bashamboo et al.,12 which were all located in the hinge region (amino acids 95–225) or the N-terminal portion of the ligand-binding domain, one of the mutation described herein was found in the hinge region (P.Gly165Arg, case 1) and two mutations (P.Asp257Asn, cases 2, and P.Ile323Thr, case 3) in the ligand-binding domain of SF1 protein. Concerning the mutation p.Gly165Arg, the possibility cannot be ruled out that a mutation in another gene either caused or contributed to the phenotype of complete meiotic arrest in the carrier. Although synonymous mutations were found more often in patients than controls, their pathogenicity currently remains unclear and these might well be rare benign variants.
Overall, the mutation frequency in Caucasian men was around or below 1% depending on subgroups analysed, which was markedly lower than the 4% reported by Bashamboo et al.12 In the French study, men of mixed ancestry were analysed and because all seven mutation carriers were of non-Caucasian extraction (three Congolese, one Tunesian, Sri Lankan, French–Vietnamese and Egyptian each), the different populations under investigation likely explain the differences in carrier frequencies. Indeed we, too, found one mutation in one of the few men of non-Caucasian ancestry, namely of Pakistani origin.
As only three probably causative NR5A1 mutations were identified, no genotype–phenotype correlation could be derived, but mutations were limited to infertile patients with azoo- or severe oligozoospermia. Two of these patients had not only highly elevated FSH serum levels but also borderline or increased LH levels in the presence of normal serum Testosterone hinting to a compensated Leydig cell insufficiency. Bashamboo et al.12 described a double NR5A1 mutation (c.368G>C, P.Gly123Ala and c.386C>T, P.Pro129Leu) in one man with sperm concentrations of 12 and 6 million/ml measured 2 years apart. Since sperm counts are well known to vary extensively also intra-individually and differences up to 50% are the norm rather than the exception,17 the assumption that sperm counts were declining in this man should be regarded with caution. All other mutation carriers in the French study had sperm concentration below 0.8 million/ml. However, sperm concentrations above 1 million/ml seem compatible with this specific NR5A1 mutation and setting a screening threshold for patients seems difficult at present. However, with a threshold of 1 million/ml, all mutations reported herein and 6 out of 7 from Bashamboo et al.12 would have been found in infertile patients.
One relatively small study found the polymorphism c.437G>C (P.Gly146Ala) significantly more frequently in 72 Japanese boys with cryptorchidism (29.2% C-allele frequency) compared with controls (18.8%, P=0.015).18 While carriers of p.Gly146Ala had varying spermatogenic phenotypes in our study, the polymorphism was exclusively found in patients without a history of cryptorchidism. We could, therefore, not confirm an association of NR5A1 P.Gly146Ala with cryptorchidism in our larger study group.
By analysing NR5A1 in a large group of almost 500 infertile males, we provide further evidence that mutations altering SF1 protein function may cause spermatogenic failure, broadening the phenotypic spectrum of NR5A1 mutations. In addition to the original study by Bashamboo et al.12, we show that this association is present also in German/Caucasian men, albeit at lower frequency. No andrological phenotype distinguishes mutation carriers in infertile but otherwise healthy men. Therefore, only screening of all men with low sperm concentrations seems currently possible to gather more data on the frequency and effects of NR5A1 mutations in infertile males. At present, it is also not possible to predict the phenotypes caused by NR5A1 mutations in offspring, for example, conceived by intracytoplasmic sperm injection, but this will be the clinically most relevant task for the future.
References
van Golde RJ, van der Avoort IA, Tuerlings JH et al: Phenotypic characteristics of male subfertility and its familial occurrence. J Androl 2004; 25: 819–823.
Yan W : Male infertility caused by spermiogenic defects: lessons from gene knockouts. Mol Cell Endocrinol 2009; 306: 24–32.
Schultz N, Hamra FK, Garbers DL : A multitude of genes expressed solely in meiotic or postmeiotic spermatogenic cells offers a myriad of contraceptive targets. Proc Natl Acad Sci USA 2003; 100: 12201–12206.
Huang WJ, Yen PH : Genetics of spermatogenic failure. Sex Dev 2008; 2: 251–259.
McLachlan RI, O’Bryan MK : State of the art for genetic testing of infertile men. J Clin Endocrinol Metab 2010; 95: 1013–1024.
Tüttelmann F, Nieschlag E : Classification of Andrological Disorders.; in Nieschlag E, Behre HM, Nieschlag S, (eds): Andrology: Male Reproductive Health and Dysfunction. Springer: Heidelberg, 2010, pp 87–92.
Tüttelmann F, Werny F, Cooper TG, Kliesch S, Simoni M, Nieschlag E : Clinical experience with azoospermia: aetiology and chances for spermatozoa detection upon biopsy. Int J Androl 2011; 34: 291–298.
Simoni M, Tüttelmann F, Gromoll J, Nieschlag E : Clinical consequences of microdeletions of the Y chromosome: the extended Münster experience. Reprod Biomed Online 2008; 16: 289–303.
Tüttelmann F, Rajpert-De Meyts E, Nieschlag E, Simoni M : Gene polymorphisms and male infertility - a meta-analysis and literature review. Reprod Biomed Online 2007; 15: 643–658.
Ferlin A, Raicu F, Gatta V, Zuccarello D, Palka G, Foresta C : Male infertility: role of genetic background. Reprod Biomed Online 2007; 14: 734–745.
Nuti F, Krausz C : Gene polymorphisms/mutations relevant to abnormal spermatogenesis. Reprod Biomed Online 2008; 16: 504–513.
Bashamboo A, Ferraz-de-Souza B, Lourenco D et al: Human male infertility associated with mutations in NR5A1 encoding steroidogenic factor 1. Am J Hum Genet 2010; 87: 505–512.
Luo X, Ikeda Y, Parker KL : A cell-specific nuclear receptor is essential for adrenal and gonadal development and sexual differentiation. Cell 1994; 77: 481–490.
Ferraz-de-Souza B, Lin L, Achermann JC : Steroidogenic factor-1 (SF-1, NR5A1) and human disease. Mol Cell Endocrinol 2011; 336: 198–205.
Tüttelmann F, Luetjens CM, Nieschlag E : Optimising workflow in andrology: a new electronic patient record and database. Asian J Androl 2006; 8: 235–241.
World Health Organization: WHO Laboratory Manual for the Examination and Processing of Human Semen. Geneva: World Health Organization, 2010.
Keel BA : Within- and between-subject variation in semen parameters in infertile men and normal semen donors. Fertil Steril 2006; 85: 128–134.
Wada Y, Okada M, Fukami M, Sasagawa I, Ogata T : Association of cryptorchidism with Gly146Ala polymorphism in the gene for steroidogenic factor-1. Fertil Steril 2006; 85: 787–790.
Acknowledgements
We thank the patients who participated and physicians who took care for them at the Centre of Reproductive Medicine and Andrology, Münster. We gratefully acknowledge the technical assistance of Anne-Lena Bröcher. We thank Stephan Köhnemann for counselling on kinship analyses. The study was supported by the Deutsche Forschungsgemeinschaft Research Unit ‘Germ Cell Potential’ (FOR 1041, grant TU 298/1–1 and TU 298/1-2 to FT and AR).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Röpke, A., Tewes, AC., Gromoll, J. et al. Comprehensive sequence analysis of the NR5A1 gene encoding steroidogenic factor 1 in a large group of infertile males. Eur J Hum Genet 21, 1012–1015 (2013). https://doi.org/10.1038/ejhg.2012.290
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2012.290
Keywords
This article is cited by
-
Pubertal development in 46,XY patients with NR5A1 mutations
Endocrine (2022)
-
Analysis of STAG3 variants in Chinese non-obstructive azoospermia patients with germ cell maturation arrest
Scientific Reports (2021)
-
Iprodione and/or chlorpyrifos exposure induced testicular toxicity in adult rats by suppression of steroidogenic genes and SIRT1/TERT/PGC-1α pathway
Environmental Science and Pollution Research (2021)
-
Disorders of spermatogenesis
Medizinische Genetik (2018)
-
Stability and Pharmacological Effects of Gene-Recombinant Wild Type and Mutant Human Adrenocorticotropic Hormone
Pharmaceutical Research (2017)
