
Abstract
Multiple endocrine neoplasia type 1 (MEN1) is an autosomal-dominant cancer syndrome that is caused by a germline mutation in the MEN1 gene encoding a tumour-suppressor protein, menin. MEN1 causes a combination of endocrine tumours such as parathyroid adenomas, pituitary adenomas, glucagonomas, gastrinomas, insulinomas, adrenocortical adenomas and non-endocrine tumours. We here present a large MEN1 family where the carriers developed mild hyperparathyroidism, multiple well-differentiated functionally active neuroendocrine tumours of the pancreas and no pituitary tumour. The causal mutation is a new double substitution in the coding region of exon 2 in the MEN1 gene c.[428T>A; 429C>T], p.Leu143His. This new mutation in the MEN1 gene is clinically relevant leading to a limited penetrance and specific phenotype.
Similar content being viewed by others

Introduction
Diagnosis of multiple endocrine neoplasia type 1 (MEN1; OMIM no. 131100) is raised by the combination of endocrine tumours such as parathyroid adenomas, pituitary adenomas, glucagonomas, gastrinomas, insulinomas, adrenocortical adenomas and non-endocrine tumours. MEN1 is an autosomal-dominant cancer disorder that is mostly caused by germline mutations in the MEN1 gene on chromosome 11q13 encoding the menin, tumour-suppressor protein.1 The somatic loss of heterozygosity is a crucial step in the MEN1 tumorigenesis. Recently, CDKN1b and AIP genes have been demonstrated to be involved in MEN1 but MEN1 gene mutations remain the main cause of the MEN1 disorder.2
Today, >1000 different MEN1 germline mutations have been reported, which are scattered over the entire gene (The Human Gene Mutation Database http://www.hgmd.cf.ac.uk).3 There is actually no consensus about a genotype–phenotype correlation.3 MEN1 gene mutations responsible for MEN1 are also involved in different variant diseases such as the familial isolated hyperparathyroidism (FIHP).4 Some MEN1 mutations were reported to have a low penetrance.5, 6 This variability in phenotypic expression could be explained by the existence of different modifier genes.7 Recently, authors argued that the stability of the mutant menin could correlate with the clinical expression of MEN1.8 The MEN1 mutations associated with FIHP were as stable or as slightly less stable than the wild-type menin and showed normal interaction with JunD, one of the interacting proteins of menin.8
A majority of MEN1 gene mutation carriers (up to 99%) will express at least one manifestation of the disease before the age of 50.9 Primary hyperparathyroidism (pHPT) is the most frequent expression of MEN1 with a penetrance between 73 and 100% by age 50 years.10, 11 The penetrance of the pancreatic neuroendocrine tumours (pNETs) is very high, reaching between 65 and 85% at the age of 60.11, 12 The pNETs arising from pancreatic islet cells may be functioning, with production of hormones such as gastrin, insulin, vasoactive intestinal polypeptide (VIP), glucagon and somatostatin, or non-functioning with the release of pancreatic polypeptide (PP).11 The prevalence of pituitary tumours (PITs) in MEN1 patients vary from 20 to 65%.13 The MEN1 gene mutation carriers still show high morbidity and mortality rates with the first cause of death being malignant pNETs and thymic NETs.14, 15
Case report
We here present a large MEN1 family where the diagnosis was made from the proband on the basis of the MEN1 criteria: the presence of two of the three main related MEN1 tumours pHPT, pNET or PIT.9 The proband manifested at 29 years diarrhoea, polyuria, polydipsia, tiredness, loss of weight and diffuse muscle pain. Her biology consisted of TSH, 1.37 mIU/l (normal range 0.4–4); fT4, 10.4 ng/l (9.3–17); PRL, 360 mIU/l (102–496); ACTH, 48.4 ng/l (7–63); cortisol, 70.8 μg/l (62–194); gastrin, 27.8 ng/l (<100); fasting insulin, 8.8 mIU/l (2–17); fasting glucagon, 76 ng/l (40–130); fasting glucose, 146 mg/dl (70–100); fasting C-peptide, 2.1 μg/l (0.4–4); fasting IGF-1, 268 μg/l (87–238); calcium, 10.5 mg/dl (8.6–9.8); phosphate, 2.1 mg/dl (2.7–4.5); PTH, 47.2 ng/l (15–65); 25-OH Vitamin D, 26 μg/l (16–52); VIP, 20 pg/ml (15–65) and chromogranine, 131 μg/l (40–170). A caudal pancreas cyst (diameter 4 cm) was detected at the MRI of the abdomen. She was treated by distal pancreatectomy and the anatomopathological examination revealed multiple PP-positive pNETs (at least six pNETs, the largest diameter was of 15 mm) with obstruction of the Wirsung canal. Her familial history revealed a suspicion of pancreatic cancer for her grandfather. During her hospitalisation, a mild hypercalcaemia (10.5 mg/dl) and PTH within reference range were detected. No hypertrophy of the parathyroid glands was found. We concluded at an early stage of pHPT. Diabetes mellitus was diagnosed, and could be kept under control with a dietary intervention. No PIT or adrenal tumour was detected. A MEN1 syndrome was suspected and molecular analysis of the MEN1 gene was performed. A putative novel DNA variant was identified, in heterozygous form, consisting of a double substitution in the coding region of exon 2 at position 428 where a thymidine was exchanged for an adenosine and at position 429 where a cytosine was exchanged for a thymidine c.[428T>A; 429C>T], leading to leucine for histidine substitution at the amino-acid level (p.Leu143His; reference sequence NM_000244). This variant was not found within a group of 100 control DNA samples.
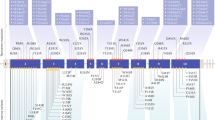
The proband has eight brothers and sisters, the parents were not alive any more (see Figure 1). All family members were tested for the novel MEN1 variant. Among the carriers (n=8), three developed mild pHPT (3/8), two multiple pNETs (2/8), one a small stable pancreas corpus cyst (diameter <1 cm; 1/8) and one an adrenal adenoma (1/8). The age range of the first symptoms was from 26 to 54 years (see Figure 1). The patients carrying the mutation were since May 2009 tested biannually by blood sampling and every 2 years by imaging (see Table 1).

Genealogical tree of the reported MEN1 family and family member’s data.
A sister of the proband carrying the putative MEN1 causal mutation developed at 54 years multiple pNETs (at least six pNETS, the largest diameter was of 12 mm), which were PP and VIP positive. She underwent a distal pancreatectomy and no adjuvant therapy was given. Further, she presented an adrenal adenoma (with a stable diameter of 5 mm) and a mild hypercalcaemia with a moderate pHPT. No PIT was detected. Two years after the pancreatectomy, she presented persistent diarrhoea with an elevated chromogranine A. The MRI of the abdomen, the endoscopic ultra sounds and the octreotide scintigraphy were negative. A next control of blood sampling and imaging is planned within 6 months to exclude a recurrence of pNETs.
No affected family member developed a PIT. Remarkably, hypercalcaemia was limited or even absent, even at advanced age (see Figure 1), and no family members underwent parathyroidectomy.
Discussion
To our knowledge, this putative novel mutation (p.Leu143His) has not been described before and we did not find it within a group of 100 control samples. To date, >1300 MEN1 mutations have been reported.3 Missense mutations account for 20% of the MEN1 mutations, the rest consists of nonsense mutations, splice-site mutations, frame shift and in-frame deletions or insertions, and whole-gene deletions.3 Neither mutation hot spot in the MEN1 gene nor phenotype–genotype correlation has been established in MEN1 disorder.3
Recently, the complex structure of menin was shown to contain a pocket that binds multiple proteins such as JunD or the oncogenic factor MLL1.16 Mutations in this pocket region could modify the menin interactions with these transcription factors and partly explain specific MEN1 phenotypes.16, 17 This putative novel mutation in exon 2 of the MEN1 gene replaces the leucine residue at position 143 with a histidine residue, a positively charged amino acid (p.Leu143His). This position is highly conserved and lies in the N-terminal α-helical bundle, which with the central domain of the menin were shown to be critical for its interaction with JunD.16 We postulate that this new double substitution MEN1 mutation affects the interaction of menin with MLL and JunD, and is clinically relevant leading up to now to a specific phenotypic spectrum of MEN1 with a limited penetrance.
The carriers who expressed the disease had mild hyperparathyroidism even at age >50 years, VIP- and PP-positive pNETs and no PIT. Some carriers even at age >50 years did not express the disease until now. Two carriers were diagnosed with pNETs at 29 years of age and at 54 years, respectively, which is still compatible with the previously reported prevalence. Indeed, MEN1 patients have a lifetime risk estimated around 75% to develop NETs of foregut origin: thymus, stomach duodenum and pancreas.10, 18
The anatomopathology showed for both patients multiple foci of pNETs with combined positive markers for PPomas and VIPomas signing a rare type of pNETs.
Remarkably, no MEN1 variant carrier had PIT, which is still compatible with the prevalence of PITs in MEN1 patients (20–65%) while most of the MEN1 patients are usually diagnosed with a prolactinoma in the fourth decade.10, 19 One carrier had an adrenal adenoma. Most of the MEN1-associated adrenal adenomas are reported to be non-secreting and stable during follow-up.20
Conclusions
We present a new double substitution mutation in exon 2 at a highly conserved position in the MEN1 gene associated with a specific clinical presentation. The carriers of this novel MEN1 mutation developed pNETs, with mild and even absent pHPT. This could indicate a limited penetrance of this novel mutation, as most carriers of MEN 1 mutations present with hypercalcaemia by the age of 50 years. Moreover, no proband developed a PIT today. Because of this new mutation, the mutant menin protein is expected to show a disturbed activity. Studies of the activity of the mutant protein could clarify its biological function and phenotypic implications.
Genetic testing allowed identification of asymptomatic carriers, and the early diagnosis of MEN1-associated pNETs in one proband. The close follow-up of MEN1 patients is known to improve clinical outcome and to reduce the mortality rate.18 Longer follow-up of carriers of this novel MEN1 mutation will allow further characterisation of this phenotype and may contribute to the genotype–phenotype discussion.
References
Piecha G, Chudek J, Wiecek A : Multiple endocrine neoplasia type 1. Eur J Intern Med 2008; 19: 99–103.
Belar O, De la Hoz C, Pérez-Nanclares G, Castaño L, Gaztambide S, Spanish MEN1 Group: Novel mutations in Men1, CDKN1b and AIP genes in patients with multiple endocrine neoplasia type 1 syndrome in Spain. Clin Endocrinol 2012; 76: 719–724.
Lemos MC, Thakker RV : Multiple endocrine neoplasia type 1 (MEN1): analysis of 1336 mutations reported in the first decade following identification of the gene. Hum Mutat 2008; 29: 22–32.
Pannett AA, Kennedy AM, Turner JJ et al: Multiple endocrine neoplasia type 1 (MEN1) germline mutations in familial isolated primary hyperparathyroidism. Clin Endocrinol 2003; 58: 639–646.
Dreijerink KM, van Beek AP, Lentjes EG et al: Acromegaly in a multiple endocrine neoplasia type 1 (MEN1) family with low penetrance of the disease. Eur J Endocrinol 2005; 153: 741–746.
Drori-Herishanu L, Horvath A, Nesterova M et al: An Intronic mutation is associated with prolactinoma in a young boy, decreased penetrance in his large family, and variable effects on MEN1 mRNA and protein. Horm Metab Res 2009; 41: 630–634.
Lemos MC, Harding B, Reed AA et al: Genetic background influences embryonic lethality and the occurrence of neural tube defects in Men1 null mice: relevance to genetic modifiers. J Endocrinol 2009; 203: 133–142.
Shimazu S, Nagamura Y, Yaguchi H, Ohkura N, Tsukada T : Correlation of mutant menin stability with clinical expression of multiple endocrine neoplasia type 1 and its incomplete forms. Cancer Sci 2011; 102: 2097–2102.
Trump D, Farren B, Wooding C et al: Clinical studies of multiple endocrine neoplasia type 1 (MEN1). QJM 1996; 89: 653–669.
Carty SE, Helm AK, Amico JA et al: The variable penetrance and spectrum of manifestations of multiple endocrine neoplasia type 1. Surgery 1998; 124: 1106–1113.
Tsukada T, Nagamura Y, Ohkura N : MEN1 gene and its mutations: basic and clinical implications. Cancer Sci 2009; 100: 209–215.
Barakat MT, Meeran K, Bloom SR : Neuroendocrine tumours. Endocr Relat Cancer 2004; 11: 1–18.
Pieterman CR, Vriens MR, Dreijerink KM, van der Luijt RB, Valk GD : Care for patients with multiple endocrine neoplasia type 1: the current evidence base. Fam Cancer 2011; 10: 157–171.
Geerdink EA, Van der Luijt RB, Lips CJ : Do patients with multiple endocrine neoplasia syndrome type 1 benefit from periodical screening? Eur J Endocrinol 2003; 149: 577–582.
Goudet P, Murat A, Binquet C et al: Risk factors and causes of death in MEN1 disease. A GTE (Groupe d’Etude des Tumeurs Endocrines) cohort study among 758 patients. World J Surg 2010; 34: 249–255.
Murai MJ, Chruszcz M, Reddy G et al: Crystal structure of menin reveals binding site for mixed lineage leukemia (MLL) protein. J Biol Chem 2011; 286: 31742–31748.
Huang J, Gurung B, Wan B et al: The same pocket in menin binds both MLL and JUND but has opposite effects on transcription. Nature 2012; 482: 542–546.
Pieterman CR, Schreinemakers JM, Koppeschaar HP et al: Multiple endocrine neoplasia type 1 (MEN1): its manifestations and effect of genetic screening on clinical outcome. Clin Endocrinol 2009; 70: 575–581.
Jensen RT, Berna MJ, Bingham DB, Norton JA : Inherited pancreatic endocrine tumor syndromes: advances in molecular pathogenesis, diagnosis, management, and controversies. Cancer 2008; 113: 1807–1843.
Langer P, Cupisti K, Bartsch DK et al: Adrenal involvement in multiple endocrine neoplasia type 1. World J Surg 2002; 26: 891–896.
Acknowledgements
We thank the technical staff of the Department of Medical Genetics of the UZ Brussel, Brussels, Belgium, for their help in the laboratory work. We also thank Olivier Vandenberg for his critical comments.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Ullmann, U., Unuane, D., Velkeniers, B. et al. A new double substitution mutation in the MEN1 gene: a limited penetrance and a specific phenotype. Eur J Hum Genet 21, 695–697 (2013). https://doi.org/10.1038/ejhg.2012.241
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2012.241
