
Abstract
Telomeres are nucleoprotein complexes that play essential roles in protecting chromosome ends. Mammalian telomeres consist of repetitive DNA sequences bound by the shelterin complex. In this complex, the POT1-TPP1 heterodimer binds to single-stranded telomeric DNAs, while TRF1 and TRF2-RAP1 interact with double-stranded telomeric DNAs. TIN2, the linchpin of this complex, simultaneously interacts with TRF1, TRF2, and TPP1 to mediate the stable assembly of the shelterin complex. However, the molecular mechanism by which TIN2 interacts with these proteins to orchestrate telomere protection remains poorly understood. Here, we report the crystal structure of the N-terminal domain of TIN2 in complex with TIN2-binding motifs from TPP1 and TRF2, revealing how TIN2 interacts cooperatively with TPP1 and TRF2. Unexpectedly, TIN2 contains a telomeric repeat factor homology (TRFH)-like domain that functions as a protein-protein interaction platform. Structure-based mutagenesis analyses suggest that TIN2 plays an important role in maintaining the stable shelterin complex required for proper telomere end protection.
Similar content being viewed by others


Introduction
Telomeres are specialized nucleoprotein complexes at the ends of linear chromosomes that play important roles in maintaining genome stability and faithfully transmitting of genetic information1. Mammalian telomeres are composed of repetitive TTAGGG DNA sequences and are bound by a six-protein shelterin complex consisting of TRF1, TRF2, RAP1, TIN2, TPP1, and POT12. Telomere uncapping by deleting shelterin components activates inappropriate DNA damage signaling and repair pathways, leading to chromosome fusion or telomere loss1,3. TRF1 and TRF2 bind to the double-stranded (ds) DNA regions of telomeres and function as docking sites for many telomere-accessory proteins4,5,6. The POT1-TPP1 heterodimer binds to the single-stranded (ss) telomeric overhangs and participates in telomere length control by regulating telomerase recruitment and processivity7,8,9. POT1-TPP1 is also required to repress an ATR-dependent DNA damage response (DDR) at telomeres10,11,12,13,14,15. TIN2 occupies a central position in this complex and simultaneously interacts with TRF1, TRF2, and TPP1, bridging three major DNA-binding modules in a highly organized manner16,17,18,19.
As the central hub of the shelterin complex, TIN2 plays critical roles in both telomere maintenance and end protection17,18,19,20,21,22,23. TIN2 is involved in TRF1-mediated telomere length regulation, stabilizing TRF1 through two distinct mechanisms. First, TIN2 protects TRF1 from tankyrase 1-mediated poly(ADP-ribosyl)ation, in turn stabilizing TRF1's association with telomeres24. Second, TIN2 competes with SCFFBX4 for TRF1 binding, thus preventing TRF1 from ubiquitin-dependent proteolysis25. TIN2 is also involved in telomere protection. By linking the POT1-TPP1 heterodimer on the 3′ ss overhang to the rest of the shelterin complex, TIN2 helps repress ATR signaling22. In addition, TIN2 inhibits ATM signaling at telomeres by stabilizing TRF2 on ds telomeric DNAs19,22. Thus, loss of TIN2 initiates DDRs via both pathways, highlighting the importance of TIN2 in telomere end protection.
Given that TIN2 interacts with three shelterin proteins and TIN2 deletion compromises the stability of both TRF1 and TRF2 at telomeres17,20,22, it is difficult to define the precise roles of each of the TIN2-mediated interactions in telomere protection. This problem is partially solved by designing TIN2 mutants that specifically disrupt its interaction with TRF119 or with TPP118. However, the importance of the TIN2-TRF2 interaction remains unclear.
With the exception of a short C-terminal TRFH-binding motif of TIN2 that has been structurally characterized4, no structural information is available for TIN2. How TIN2 functions as a hub to bridge the ds and ss telomeric regions remains elusive. Here, we determine the structure of TIN2-TPP1-TRF2 ternary complex and demonstrate how TIN2 interacts cooperatively with TPP1 and TRF2. Interestingly, N-terminal domain of TIN2 shows a striking structural similarity to the TRFH domains of TRF1 and TRF2, suggesting that they may have a common ancestor, despite having diverged functionally during evolution. Our structural studies enable us to generate point mutations that specifically disrupt TIN2's capacity to interact with TRF1, TRF2, or TPP1. We found that the TIN2-TPP1 interaction is important to repress alternative non-homologous end joining (A-NHEJ)-mediated chromatid and sister telomere fusions, while the TIN2-TRF2 and TIN2-TRF1 interactions are required to prevent both classical-NHEJ (C-NHEJ)-mediated chromosome fusions and A-NHEJ-mediated chromatid fusions.
Results
Structure of the TIN2-TPP1-TRF2 ternary complex
Previous studies have shown that TIN2 interacts with TRF2 via two interacting modules4. One is the short TRFH-binding motif (TBM) (residues 256-276) at the C-terminal portion of TIN2, which interacts with both TRF1TRFH and TRF2TRFH (Figure 1A)4. The other is the N-terminal domain of TIN2 (residues 2-202), which recognizes a short TIN2-binding motif of TRF2 (TRF2TBM, residues 350-366) (Figure 1A). Here, we used microscale thermophoresis (MST) to evaluate the contribution of each TIN2 module to the TIN2-TRF2 interaction (Supplementary information, Figure S1A). We found that TIN22-202 binds to TRF2FL (full-length TRF2) with a Kd of 1.5 μM, nearly identical to the affinity of full-length TIN2 to TRF2FL (Supplementary information, Figure S1A). In contrast, the C-terminal half of TIN2 (residues 200-355) interacts with TRF2FL with a much lower affinity (Kd∼29 μM). This result confirmed that TIN2 has two TRF2-binding modules and the N-terminal region of TIN2 is the dominant binding site for TRF2FL. Next, we explored the interaction between TIN2 and TPP1. The C-terminal region of TPP1 (residues 480-544) has been shown previously to bind TIN221,26. Further mapping by yeast two-hybrid analysis revealed that a TPP1 fragment consisting of residues 510-544 was both necessary and sufficient for interaction with TIN2 (Supplementary information, Figure S1B). Hereafter, we will refer to TPP1510-544 as the TIN2-binding motif of TPP1 (TPP1TBM) (Figure 1A).

Structure of the TIN2TRFH-TPP1TBM-TRF2TBM complex. (A) Domain organization of the TIN2, TPP1, and TRF2. OB, oligosaccharide-binding fold; PBM in TPP1, POT1-binding motif; TBM in TPP1, TIN2-binding motif; TBM in TIN2, TRFH-binding motif; DC, dyskeratosis congenita hotspot; Basic, an N-terminal basic-residue-rich domain; TRFH, telomeric repeat factor homology domain; RBM in TRF2, RAP1-binding motif; TBM in TRF2, TIN2-binding motif; Myb, a C-terminal DNA-binding domain. (B) Two orthogonal views of the overall structure of the TIN2TRFH-TPP1TBM-TRF2TBM complex. The left panel is shown in cartoon diagram, and TIN2TRFH in the right panel is shown in surface representation. TIN2TRFH is colored in green (helical bundle involved in TRF2 binding) and cyan (helical bundle involved in TPP1 binding). TPP1TBM and TRF2TBM are colored in yellow and magenta, respectively. (C) The brace helix α7 mediates cooperative binding with TPP1TBM and TRF2TBM. (D) TPP1TBM promotes TRF2TBM interaction with TIN2TRFH, and TRF2TBM also enhances TPP1TBM interaction with TIN2TRFH, as shown by fluorescence polarization assays.
We found that TIN22-202 can bind to both TPP1TBM and TRF2TBM simultaneously, forming a stable ternary complex (Supplementary information, Figure S1C). After extensive screenings, we successfully crystallized a complex containing all three proteins (TIN22-202, TPP1TBM, and TRF2TBM, Figure 1A) and determined its structure at a resolution of 2.2 Å by single-wavelength anomalous dispersion with selenomethionine-substituted crystals (Table 1). This structure was refined to an R-value of 19.5% (Rfree∼22.7%) with good geometry. The electron density map unambiguously allowed us to trace the majority of the complex (Supplementary information, Figure S2A and S2B).
TIN22-202 adopts a compact fold with nine α-helices tightly packed together with linear dimensions of ∼60Å × 40Å × 30Å (Figure 1B). Residues 91-96 of TIN2 are not modeled in the structure due to their poor electron density, which is consistent with the observation that this short fragment of TIN2 is variable in size across species (Supplementary information, Figure S2C). Each TIN22-202 molecule binds to one TRF2TBM and to one TPP1TBM using distinct surfaces (Figure 1B). TIN22-202 can be roughly divided into two helix bundles. Helices α3, α4, α5, α6, and the N-terminal half of α7 (α7a) of TIN22-202 form the first helical bundle, packing against TRF2TBM (Figure 1B). The other is comprised of helices α1, α2, α8, α9, and the C-terminal half of α7 (α7b) and this helical bundle interacts with TPP1TBM (Figure 1B).
Previous studies revealed that TPP1 promotes the interaction between TIN2 and TRF221. The ternary complex structure shown here suggests a molecular mechanism for this cooperativity. Although TPP1TBM and TRF2TBM bind to two different pockets in TIN22-202, the long helix 7 of TIN22-202 contacts both TPP1TBM and TRF2TBM simultaneously (Figure 1C). This α7 helix functions like a seesaw, with its two ends seated by TPP1TBM and TRF2TBM, respectively (Figure 1C). We postulate that the binding of TPP1TBM to TIN22-202 will allosterically regulate the α7 helix of TIN2 to maintain an optimal configuration to bind to TRF2TBM, and vice versa. In support of this notion, fluorescence polarization assays confirmed that the binding affinity between TIN22-202-TPP1TBM and TRF2TBM is ∼2.6-fold higher than that between TIN22-202 and TRF2TBM (Figure 1D), indicating that TPP1TBM can enhance the interaction between TIN22-202 and TRF2TBM. Similarly, TRF2TBM binding to TIN22-202 also increased the binding affinity between TIN22-202 and TPP1TBM by 3.7-fold (Figure 1D). Taken together, these data suggest that TIN22-202 cooperatively interacts with TPP1 and TRF2 to ensure the stable assembly of the shelterin complex.
TIN22-202 structurally resembles the TRFH domains of TRF1 and TRF2
Next, we performed an unbiased search for structurally homologous proteins to the TIN22-202 domain using DALI27. Surprisingly, we found that TIN22-202 is closely related to the TRFH domains of TRF1 and TRF2 (Figure 2A and 2B). TIN22-202 can be superimposed onto the TRF1TRFH and TRF2TRFH domains with root-mean-square deviation (rmsd) values of 3.5 and 3.4 Å for 123 and 131 equivalent Cα pairs, respectively, although structure-based sequence alignment shows <9% identity between TIN22-202 and TRFH domains (Figure 2C). Superimposition of TIN22-202 with TRF1TRFH and TRF2TRFH clearly reveals that the first seven α-helices of the TIN22-202 structurally resemble the α3-α9 helices in TRF1/2TRFH (Figure 2A and 2C). In addition, both TIN22-202 and the TRFH domains of TRF1 and TRF2 function as protein-interaction platforms that utilize a similar surface to interact with their respective binding partners — TPP1 for TIN22-202 and TBM-containing proteins for TRF1TRFH and TRF2TRFH (Figure 2B). Together, these structural and functional similarities suggest that TIN22-202 and the TRFH domains of TRF1 and TRF2 are very likely to be evolutionarily related. Therefore, we name TIN22-202 as the TRFH domain of TIN2 (TIN2TRFH) (Figure 1A).

TIN2TRFH contains a TRFH-like fold. (A) Superimposition of TIN2TRFH (green), TRF1TRFH (light blue) and TRF2TRFH (blue) shown in two orthogonal views. The left panel shows monomeric TRF1/2TRFH and the right panel shows the dimeric form of TRF1/2TRFH with one monomer colored in gray. TIN2TRFH α8 and α9 disrupt the dimerization interface formed by α1, α2, and α10 in TRF2TRFH. (B) Superimposition of TIN2TRFH-TPP1TBM-TRF2TBM and TRF2TRFH-ApolloTBM. TPP1TBM and ApolloTBM bind to the similar surface pockets on TIN2TRFH and TRF2TRFH, respectively. (C) Structure-based sequence alignment of TIN2TRFH, TRF1TRFH, and TRF2TRFH. Secondary structure assignments based on the TIN2TRFH and TRF2TRFH structures are shown as cylinders (α-helices) and lines (loops).
The TIN2-TPP1 interface
In the ternary complex, each TPP1TBM polypeptide is folded into a helix-loop-helix motif (H1-L12-H2) (Figure 1B). Both helices and the connecting loop make extensive contacts with TIN2TRFH, burying ∼1 268 Å2 of surface area at their interface (Figure 1B). The driving force for the binding of TPP1TBM to TIN2TRFH is van der Waals interactions, as most conserved residues of TPP1TBM are hydrophobic in nature (Supplementary information, Figure S2D). This observation is consistent with the finding that TPP1TBM cannot be purified by itself and must be coexpressed and copurified along with TIN2TRFH. The H1 and H2 helices of TPP1TBM pack against the floors of two hydrophobic grooves on TIN2TRFH, separated by helices α1 and α2 of TIN2 (Figure 3A and 3B). In addition, the loop connecting helices H1 and H2 of TPP1TBM also makes intimate contacts with TIN2TRFH (Figure 3A). The core of this extended interface between TPP1TBM and TIN2TRFH consists of a panel of hydrophobic residues from both proteins, including V515, L520, L524, M525, W527, and L529 of TPP1TBM, and V6, L12, A15, A18, F37, L158, and L162 of TIN2TRFH (Figure 3A and 3B). The extensive contacts among the side chains of these residues mediate the specificity of TPP1TBM recognition by TIN2TRFH.

The interface between TIN2TRFH and TPP1TBM. (A) Details of hydrophobic interactions around H1 and L12 of TPP1. The interaction residues are presented as ball-and-stick models. TPP1 residues are colored in yellow and TIN2 residues are colored in cyan. (B) Details of hydrophobic contacts between H2 of TPP1 and TIN2. (C, D) Effects of mutations in the TPP1TBM (C) and TIN2TRFH (D) domains on the interaction between TIN2 and TPP1 analyzed in a yeast two-hybrid assay. Data are the average of three independent β-galactosidase measurements.
To validate the TIN2-TPP1 interface observed in the crystal structure, we mutated specific residues involved in the TIN2-TPP1 interaction and examined their binding using a yeast two-hybrid assay. Consistent with the structural data, mutations of single residues from helices H1, H2, or loop L12 of TPP1TBM destabilized the TPP1-TIN2 interaction, suggesting that all three modules contribute to TIN2-TPP1 interaction (Figure 3C). In particular, mutations on the H2 helix of TPP1TBM conferred the most disrupting effects, suggesting that helix H2 is the most important region responsible for binding with TIN2 (Figure 3C). Similarly, hydrophobic residues on the TIN2 interface are also required for binding to TPP1. Substitution of TIN2A15 with a positively charged, bulky arginine residue completely disrupted the TIN2-TPP1 interaction (Figure 3D). In contrast, alanine substitution of TIN2 interface residues did not alter the hydrophobicity of the binding pocket for TPP1TBM, and thus only partially impeded the interaction (Figure 3D). Taken together, we conclude that the hydrophobic contacts observed in the crystal structure are the major biophysical driving force for the interaction between TIN2 and TPP1.
The TIN2-TRF2 interface
The interaction between TIN2TRFH and TRF2TBM is mediated by a combination of van der Waals contacts and electrostatic interactions, burying ∼740 Å2 of surface area that corresponds to 47% of the total surface area of TRF2TBM. Close inspection of the TIN2TRFH-TRF2TBM interface reveals three adjacent binding modules (Figure 4A; Supplementary information, Figure S2E). The most prominent contribution to the TIN2-TRF2 interface is from the central helix of TRF2TBM (359ISRLVL364) (Figure 4A). Three hydrophobic residues (TRF2I359, TRF2L362, and TRF2V363) in the center of this helix form a hydrophobic core that fits snugly into a large triangular-shaped pocket formed by helices α3, α5, and α7 of TIN2TRFH (Figure 4B). The side chains of each of these residues make intimate contacts with a panel of hydrophobic amino acids at the corresponding vertices in the triangular pocket of TIN2TRFH (Figure 4B). Consistent with their importance for binding, arginine substitution of any of these residues severely impaired the interaction between TRF2 and TIN2, while substitutions with nonpolar alanine residue only modestly weakened this interaction (Figure 4E). Yeast two-hybrid analysis also revealed that TIN2 mutations on the other side of the interface (F87A, G60R, and A110R) completely disrupted the TIN2-TRF2 interaction (Figure 4B and 4F), further underscoring the importance of the hydrophobic contacts for the interaction between TRF2TBM and TIN2TRFH.

The interface between TIN2TRFH and TRF2TBM. (A) TRF2TBM sits in a groove of TIN2TRFH shown in surface representation. TIN2TRFH is colored according to its electrostatic potential (positive potential, blue; negative potential, red). (B) Details of hydrophobic interactions in the center of TRF2TBM. The interaction residues are presented as ball-and-stick models. TRF2 residues are colored in magenta and TIN2 residues are colored in green. (C) The C-terminal glutamate residues in TRF2TBM form salt bridges and hydrogen bonds with basic residues from α3 and α5 of TIN2TRFH. Salt bridges and hydrogen-bonding interactions are shown as orange dashed lines. (D) The positively charged residues in the N-terminal TRF2TBM form salt bridges and hydrogen bonds with basic residues from α5 and α6 of TIN2TRFH. (E, F) Effects of mutations in the TRF2TBM (E) and TIN2TRFH (F) domains on the interaction between TIN2 and TRF2 analyzed in a yeast two-hybrid assay. Data are the average of three independent β-galactosidase measurements.
In addition to these hydrophobic contacts, the backbone carbonyls of TRF2L362 and TRF2V363 accept two hydrogen bonds from the side chain of TIN2R56 (Figure 4C), while the backbone amide and side-chain hydroxyl group of TRF2S360 donate two hydrogen bonds to TIN2Y139 and TIN2E138 (Figure 4D). These hydrogen-bonding interactions further stabilize interactions between the TRF2TBM helix and TIN2TRFH. Moreover, both the N- and C-termini of TRF2TBM contribute to the binding to TIN2 via electrostatic interactions. They function as two arms that facilitate the docking of the central helix of TRF2TBM onto TIN2 (Figure 4A). The C-terminal tail of TRF2TBM contains two glutamate residues (365EE366), which extend into a basic patch of TIN2 (Figure 4A). TRF2E365 forms an intermolecular salt bridge with TIN2K106 and an intramolecular salt bridge with TRF2R361(Figure 4C). TRF2E366 mediates two electrostatic contacts with TIN2R56 and TIN2R52 (Figure 4C). In sharp contrast, the N-terminus of TRF2TBM (355KRMT358) is basic in nature and sits on an acidic surface of TIN2 (Figure 4A). TRF2R356 and TRF2K355 form three salt bridges with TIN2E109 and TIN2E138 (Figure 4D). Charge-swapping mutations of these residues impaired the binding of TRF2 to TIN2, underscoring the importance of these electrostatic interactions to the TIN2TRFH-TRF2TBM interface (Figure 4E). Interestingly, a TRF2R356C mutation has been identified in lung adenocarcinoma28 and a TIN2K106N mutation has been found in neuroendocrine tumors29, suggesting that mutations disrupting the TRF2-TIN2 interaction may be cancer-promoting.
Mutational analyses of TIN2-mediated interactions
To extend the results from our structural studies, we examined whether the TIN2 mutations disrupted the interactions with TRF1, TRF2, and TPP1 in vivo. We generated specific point mutations in TIN2 identified by our structural studies to disrupt its binding to only one interaction partner: A15R to disrupt the TIN2-TPP1 interaction, L260E to disrupt the TIN2-TRF1 interaction, and G60R or A110R to disrupt the TIN2-TRF2 interaction. Co-immunoprecipitation (Co-IP) analysis revealed that the A15R mutation completely abolished the interaction between TIN2 and TPP1 while not affecting TIN2-TRF2 and TIN2-TRF1 interactions (Figure 5A). The L260E mutation specifically abolished the interaction of TIN2 with Myc-TRF1 but not with TPP1 or TRF2 (Figure 5B). Notably, the G60R and A110R mutations of TIN2 severely reduced the interaction with TRF2, but did not eliminate it (Figure 5C). Combining the TIN2 mutation L260E with G60R or A110R completely disrupted the interaction with TRF2 (Figure 5C). This result indicates that the secondary TRF2-binding modules of TIN2 (TIN2TBM, residues 256-276) can mediate a weak interaction with TRF2 when the TIN2TRFH-TRF2TBM interaction is disrupted (Figure 1A; Supplementary information, Figure S1A), although we cannot exclude the possibility that the secondary weak-binding site of TIN2 (TIN2TBM) may indirectly interact with TRF2. How TIN2TBM contributes to TRF2 binding in vivo needs further investigation.

Analysis of TIN2 interaction with binding partners in vivo. (A-C) 293T cells expressing the indicated protein were immunoprecipitated with Flag antibody and immunoblotted with Flag and Myc antibodies. The input represents 5% of the total cell lysate used for immunoprecipitation. GADPH or actin was used as loading control. (D) WT MEFs were reconstituted with WT or indicated mTIN2 mutants before Tin2 shRNA was used to deplete endogenous TIN2. Telomeres were visualized with telomere PNA-FISH (red), anti-Flag antibody to visualize mTIN2 (green), and DAPI staining to visualize nuclei (blue). (E) Quantification of the percent of cells with ≥5 foci of WT Flag mTIN2 and the mutants on telomeres in (D). Data are the mean of two independent experiments ± SEM; more than 100 nuclei were examined per experiment.
To confirm our Co-IP results, we investigated whether the telomeric accumulation of TIN2 depends on its interactions with TRF1, TRF2, or TPP1 in vivo. We expressed shRNA-resistant WT Flag-mTIN2 or Flag-mTIN2 mutants in mouse embryo fibroblasts (MEFs) and then removed endogenous TIN2 using shRNA against mTin2 (shTin2). Immunofluorescence microscopy (IF) revealed that ∼90% of the cells expressing both WT Flag-mTIN2 and Flag-mTIN2A15R displayed ≥5 TIN2-positive foci colocalized with telomeres per nucleus (Figure 5D and 5E), suggesting that interaction with mTPP1 is dispensable for mTIN2's localization to telomeres. In contrast, mTIN2S60R (equivalent to hTIN2G60R) and mTIN2A103R (equivalent to hTIN2A110R) mutants showed telomeric accumulation only in ∼45% of the cells, and showed diffuse nucleoplasmic staining in the remaining cells (Figure 5D and 5E). The mTIN2L247E(equivalent to hTIN2L260E) mutant was completely unable to localize to telomeres (Figure 5D and 5E). We also generated triple (Flag-mTIN2A15R, A103R, L247E) and quadruple (Flag-mTIN2A15R, S60R, A103R, L247E) mutants and, as expected, neither of them localized to telomeres (Figure 5D and 5E). These data suggest that the interaction between mTIN2 and mTRF1 is absolutely essential for mTIN2 localization to telomeres, consistent with an earlier report19. We also examined the telomeric localization of human TIN2 mutants in human U2OS cells. Similarly, we found that Flag-TIN2A15R was localized to telomeres as efficiently as TIN2WT(Supplementary information, Figure S3A and S3B). In contrast, <10% of the cells expressing TIN2G60R, TIN2A110R, and TIN2L260E mutants showed ≥5 TIN2-positive foci on telomeres (Supplementary information, Figure S3A and S3B). No colocalization of Flag-TIN2 triple and quadruple mutants with telomeres was detected (Supplementary information, Figure S3A and S3B). Taken together, these results suggest that interactions with both TRF1 and TRF2, but not TPP1, are required for efficient TIN2 localization to telomeres in human and mouse cells.
TIN2 protects telomeres from engaging in DNA damage signaling and repair
To determine whether perturbing TIN2's interaction with TRF1, TRF2, and TPP1 activates DDR at telomeres, we used the dysfunctional telomere-induced DNA damage foci (TIF) assay to monitor the recruitment of DNA damage markers γ-H2AX and 53BP1 to telomeres bearing TIN2 mutations. While TIN2-depleted MEFs reconstituted with vector control resulted in TIF formation in 40% of MEFs examined, reconstitution with WT mTIN2 almost completely repressed the localization of γ-H2AX and 53BP1 to telomeres, with only ∼10% of cells displaying ≥5 TIFs per nucleus (Figure 6A and 6B; Supplementary information, Figure S4A and S4B). In sharp contrast, reconstitution with mTIN2A15R, mTIN2S60R, mTIN2A103R, and mTIN2L247E all resulted in robust TIF formation, with 60%-80% of cells displaying ≥5 TIFs per nucleus (Figure 6A and 6B; Supplementary information, Figure S4A and S4B). This two-fold increase in the number of TIFs over vector baseline levels suggests that these single TIN2 mutants exerted a dominant negative effect at telomeres (Figure 6B; Supplementary information, Figure S4B), a notion further supported by the observation that increased TIF formation over baseline levels was not observed in MEFs expressing triple or quadruple mTIN2 mutants (Figure 6B; Supplementary information, Figure S4B). Consistent with these results, expression of single, but not triple or quadruple human TIN2 mutants in TIN2-depleted U2OS cells resulted in robust TIF formation (Supplementary information, Figure S4C and S4D). These data collectively suggest that the interactions of TIN2 with TRF1, TRF2, and TPP1 are all required to repress telomere damage signaling.

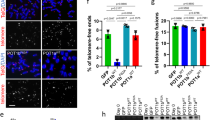
TIN2 protects telomeres from DNA damage signaling and chromosome fusions. (A) Colocalization of γ-H2AX with telomeres in reconstituted WT or indicated mTIN2 mutants. Telomeres were visualized by PNA-FISH (red), anti-γ-H2AX antibody to visualize DNA damage (green), and DAPI to visualize nuclei (blue). (B) Quantification of the percentage of cells containing ≥5 positive γ-H2AX TIFs in (A). Data represent the mean of two independent experiments ± SEM; more than 125 nuclei were analyzed per experiment. (C) MEFs expressing either WT or mTIN2 mutants were infected with either control vector or shTin2 for 120 h, after which metaphases were prepared and telomere fusions were visualized by CO-FISH. FITC-OO-(TTAGGG)4 (green) was used to visualize the leading strand and Tam-OO-(CCCTAA)4 (red) was used to visualize the lagging strand. DAPI (blue) visualized chromosomes. Arrowheads point to fused chromosomes: chromosome fusions (white arrows), and an unequal mixture of leading and lagging G2 chromosome fusions (orange arrows). (D) Quantification of chromosome-type fusions in (C). (E) Quantification of chromatid-type and sister fusions in (C). mTPP1ΔRD is a dominant-negative mutant of mTPP1 in which POT1-binding recruitment domain (RD) is deleted. Expression of mTPP1ΔRD in MEFs will remove endogenous TPP1-POT1a/b complex from telomeres. Data represent the average of two independent experiments as mean ± SEM from more than 70 metaphases examined.
To determine the types of telomere fusions observed in TIN2-deficient cells reconstituted with TIN2 mutants, we used chromosome orientation (CO)-FISH, a cytogenetics technique which can differentiate between the telomere fusions generated by leading- and lagging-strand DNA synthesis (Supplementary information, Figure S5A). Depletion of TIN2 with shRNA increased the number of chromosome and chromatid fusions, while reconstitution with WT TIN2 repressed this fusion phenotype (Figure 6C and 6D). TIN2-depleted MEFs reconstituted with mTIN2S60R, mTIN2A103R, or mTIN2L247E all displayed robust end-to-end chromosome fusions, which are ∼6-fold above fusion levels observed in cells expressing wild-type TIN2 (Figure 6C and 6D; Supplementary information, Figure S5A). Examination of fusion sites revealed that all three TIN2 mutants induced a complex mixture of chromosome- and chromatid-type fusions, including G1-type end-to-end chromosome fusions with leading and lagging telomere signal characteristic of TRF2 loss (Figure 6C–6E; Supplementary information, Figure S5A and S5B)30,31,32. In addition, we observed fusion events suggestive of postreplicative G2 repair, including chromosome fusions with unequal leading and lagging telomere signal, chromatid-type fusions involving both leading- and lagging-strand telomeres, and sister telomere fusions involving both chromatid arms reminiscent of POT1-TPP1 loss (Figure 6C, 6E; Supplementary information, Figure S5A and S5B)10,11,12,13,14,33. These results suggest that both TIN2-TRF1 and TIN2-TRF2 interactions are required to prevent the generation of both chromosome- and chromatid-type fusions. MEFs expressing mTIN2A15R also displayed an increase in the number of G1- or G2-type chromosome fusions over the background levels, similar to those observed in shTin2-treated MEFs (Figure 6C and 6D). This result suggests that the TIN2-TPP1 interaction is also important to repress chromosome fusions, although not to extent as TIN2-TRF1 and TIN2-TRF2 interactions. In addition, sister telomere fusions and chromatid fusions were abundant in MEFs expressing mTIN2A15R, suggesting that TIN2's interaction with TPP1-POT1 is also required to repress postreplicative chromatid fusion events (Figure 6C and 6E).
DNA double-strand breaks can be repaired either by the error-prone non-homologous end joining (alternative- or C-NHEJ)-mediated repair, or the error-free homology-directed repair (HDR) pathways. C-NHEJ is the major form of NHEJ and is mediated by the DNA-PK complex (DNA-PKcs, Ku70, and Ku80) and LIG4/XRCC4/XLF complex, while A-NHEJ does not require the above complexes33,34,35,36,37,38. To determine whether the TIN2 mutant-induced chromosome and chromatid fusions are a result of C- or A-NHEJ-mediated repair, we reconstituted WT or TIN2 mutants in Ku70−/− MEFs (Supplementary information, Figure S5C and S5D). Ku70 is essential for C-NHEJ-mediated repair but represses A-NHEJ-mediated repair33,34,35,36,37,38. Thus, any chromosome fusions observed in the absence of Ku70 result from A-NHEJ repair. Compared to Ku70+/+ MEFs, expression of mTIN2S60R, mTIN2A103R, or mTIN2L247E in Ku70−/− MEFs led to a decreased number of C-NHEJ-mediated chromosome fusions, while chromatid and sister fusions increased to involve over 10% of all chromatid ends (compare Supplementary information, Figure S5E–S5F with Figure 6D–6E). This result suggests that TIN2-TRF1 and TIN2-TRF2 interactions are important to repress A-NHEJ-mediated chromatid and sister fusions in addition to C-NHEJ-mediated chromosome fusions. In addition, expression of mTIN2A15R in Ku70−/− MEFs resulted in increased sister telomere fusions and chromatid-type fusions compared to WT MEFs, suggesting that the TIN2-TPP1 complex is also involved in repression of A-NHEJ-mediated chromatid and sister telomere fusions (Figure 6E; Supplementary information, Figure S5F). Taken together, our data suggest that TIN2-TRF1 and TIN2-TRF2 interactions are essential to repress telomere damage signaling, preventing both C-NHEJ-mediated chromosome fusions and A-NHEJ-mediated chromatid fusions. The TIN2-TPP1 interaction is required to repress A-NHEJ-mediated chromatid, sister telomere fusions, and to a smaller extent, C-NHEJ-mediated chromosome fusions.
Discussion
Shelterin regulates telomere maintenance and inhibits DDR at telomeres2. In the present study, we report the crystal structure of N-terminal domain of TIN2 in complex with TIN2-binding motifs from TPP1 and TRF2, which represents the central core shelterin subcomplex. Our structural analyses finally complete the high-resolution views of all building blocks of the shelterin complex (Figure 7A and 7B).

The structure of human shelterin complex. (A) Domain organizations of the shelterin components. (B) The structural model of human shelterin complex based on current available structures, including TRF1Myb-dsDNA (PDB: 1W0T), TRF2Myb-dsDNA (PDB: 1W0U), POT1OB1+OB2-ssDNA (PDB: 1XJV), TRF1TRFH in complex with TIN2TBM (PDB: 3BQO), TRF2TRFH in complex with TIN2TBM (PDB: 3BU8), RAP1BRCT (modeled from PDB 2L42), RAP1Myb(PDB: 1FEX), RAP1RCT in complex with TRF2RBM (PDB: 3K6G), TPP1OB (PDB: 2I46), POT1OB3+HJRJ in complex with TPP1PBM (PDB: 5H65), and TIN2TRFH in complex with TPP1TBM and TRF2TBM. All DNAs are shown in yellow. Dashed lines indicate the flexible linkers connecting these structural modules. For clear illustration purpose, only one RAP1 and TIN2 are presented, and only one TRF2TBM and TRF2RBM from a TRF2 monomer are shown.
Our current data and previous structural analyses suggest that all of the shelterin subcomplexes utilize a domain-peptide-interaction mechanism (Figure 7A). The TRF1/TRF2 TRFH domain recognizes a conserved TBM4, while the RCT domain of RAP1 interacts with a helical peptide of TRF2 adjacent to TRFH domain39. The POT1 C-terminal region including an OB fold and a Holliday junction-like domain binds with a long fragment of TPP140,41. Here, we show that TIN2 TRFH domain binds to short motifs from TPP1 and TRF2 to form the TIN2-TPP1-TRF2 subcomplex. This domain-peptide-interaction mechanism is also employed by telomeric protein complexes from other species, including the fission yeast SpRap1RCT-Taz1RBM complex39, the budding yeast ScRap1RCT-Sir3RBM39, and ScRap1RCT-Rif1RBM complexes42. This conserved domain-peptide-interaction mechanism provides the possibility for dynamic regulation of complex formation or dissociation. Any modification on a single residue could abolish the binding affinity between a domain and a peptide due to the relatively small contacting interface. This notion is supported by our recent work showing that the interaction between TRF2 and NBS1 is modulated by NBS1's cell-cycle-dependent phosphorylation status6. The assembly of other shelterin subcomplexes, which utilizes this domain-peptide mechanism, may also be susceptible to regulation by similar cell-cycle-dependent or DDR-dependent phosphorylation events. The structure of TIN2-TRF2-TPP1 shown here implies a potential regulatory mechanism for the stability of TIN2-TRF2 complex. Indeed, TRF2T358 in the TIN2-binding motif is known to be phosphorylated by Aurora C43. Our crystal structure reveals that TRF2T358 sits in an acidic pocket of TIN2 (Figure 4A), and thus a negatively charged phosphate group deposited on T358 would weaken or disrupt the interaction between TIN2 and TRF2. Additionally, we found that TRF2 contains a highly conserved PIKK phosphorylation site at S368, immediately following the TIN2-binding motif of TRF2 (residues 350-366). Since the C-terminus of TRF2TBM is close to a basic surface of TIN2, phosphorylation of S368 is expected to enhance the TIN2-TRF2 binding. Whether the assembly of the TIN2-TRF2-TPP1 complex is modulated by such phosphorylation events in vivo is currently being investigated.
The most striking finding in this study is that TIN2TRFH adopts a TRFH-like fold. TRFH domain was first identified in human TRF1 and TRF244,45. Despite the high degree of structural similarity, there are some differences between TIN2TRFH and the TRFH domains of TRF1 and TRF2. The TIN2TRFH does not have the equivalent α1, α2, and α10 helices in TRF1/2TRFH (Figure 2A), which are important for TRF1/2TRFH-mediated dimerization of TRF1/2 proteins44. Instead, the C-terminal α8 and α9 helices of TIN2TRFH occupy the equivalent positions of helices α1 and α2 in the TRFH domains of TRF1 and TRF2, blocking the dimerization interface found in TRF1TRFH and TRF2TRFH (Figure 2A). In agreement with this observation, TIN2TRFH exists as a monomer in solution, as revealed by gel-filtration chromatography analysis and multiangle light scattering (Supplementary information, Figures S1C and S6A). Recently, TRFH domain of TRF2 has been reported to bind and wrap DNA to regulate the topology of telomeres46. The DNA-binding activity of TRF2TRFH relies on seven lysine and two arginine residues. Structure-based alignment showed that TIN2 only has two lysine residues at the equivalent positions (Supplementary information, Figure S6B), so it is predicted that TIN2TRFH may not have the DNA-binding capacity. Electrophoresis mobility shift assays confirmed that TIN2TRFH cannot bind to dsDNA as TRF2TRFH does (Supplementary information, Figure S6C). These analyses indicate that TRFH domain might be an ancient domain in telomere-binding proteins and has gained significant structural and functional plasticity during evolution.
The TIN2-TPP1-TRF2 structural model rationalizes previously reported mutagenesis data showing that TIN2 binding to TPP1 was abolished in TIN2 mutants such as ΔN18, F37D/L38E, and L48E47, as these key residues on the α1 and α2 helices of TIN2 are required to mediate hydrophobic contacts with TPP1TBM (Figure 3A and 3B). Frescas et al.18 has generated a TIN2 mutant (TIN2Δ132-188) to specifically remove TIN2-TPP1 interaction while not affecting TIN2-TRF2 and TIN2-TRF1 interactions. TIN2132-188 contains helices α6, α7, α8, and the starting turn of α9. The helices α7, α8, and the connecting loop L78 of TIN2 make extensive contacts with TPP1TBM (Figure 3A, 3B; Supplementary information, Figure S7A), so deletion of TIN2132-188 will disrupt the interaction with TPP1. However, α6 and α7 are also involved in TRF2 interaction, so deletion of residues 132-188 might impair the interaction with TRF2 as well (Supplementary information, Figure S7A). More importantly, helices α6-α8 are the integral part of a compact helical bundle (Figure 1B), making extensive hydrophobic contacts with the rest of TIN2TRFH (Supplementary information, Figure S7B). In particular, α7 is dominantly composed of hydrophobic residues and is surrounded by a hydrophobic groove formed by α1, α2, and α3 of TIN2 (Supplementary information, Figure S7C). Thus, deletion of TIN2132-188 is expected to disrupt the overall fold of TIN2TRFH. This prediction is confirmed by the fact that the deletion of helices α8-α9 (TIN22-160 construct) cannot yield a soluble protein (Supplementary information, Figure S7D). Thus, the TIN2 mutant that lacks residues 132-188 is not a good candidate to mimic the TPP1-binding-deficient TIN2 allele. For this reason, in the present study, we designed point mutations, instead of fragment deletions, to specifically disrupt the TIN2 interactions with binding partners and to dissect the functions of TIN2 in the cells.
Our structure-based mutational analyses shed light on how specific shelterin subcomplexes repress distinct DNA damage signaling and repair mechanisms at telomeres. We found that the TIN2A15R mutant, which is unable to interact with TPP1 but localizes to telomeres through interactions with TRF2 and TRF1, induced a robust DDR, increased chromatid and sister telomere fusions, and slightly increased in chromosome fusions (Figure 6D and 6E). It is unclear whether these chromosome fusions arose from G1 fusions or postreplicative G2 fusions. End-to-end chromosome fusion can also arise from duplication of the chromatid-type fusions formed in the preceding G248. These phenotypes bear some resemblance to those observed when the POT1-TPP1 heterodimer is depleted10,11,12,13,14,33. It suggests that the major function of TIN2-TPP1 interaction is to load POT1-TPP1 heterodimer to telomeres to protect telomeres from engaging in ATR-dependent DNA damage signaling and A-NHEJ-mediated repair at telomeres, consistent with previous TIN2 knockout studies22. On the contrary, mTIN2S60R, mTIN2A103R, and mTIN2L247E mutants induced complicated phenotypes reminiscent of both TRF2 loss and POT1-TPP1 loss, including both chromosome and chromatid fusions10,11,12,30,31,33,38. This result suggests that both TIN2-TRF1 and TIN2-TRF2 interactions are required for shelterin complex stability. This is consistent with the previous report that TIN2 depletion or overexpression of TIN2-15, a dominant negative mutant which cannot interact with TRF1, destabilized both TRF1 and TRF217,20. Taken together, our mutagenesis data provided a good starting point to fully understand the functional significance of TIN2-mediated interactions in telomere protection.
In the past few decades, structural studies of shelterin components have been greatly advanced to facilitate better understanding of the molecular mechanisms of telomere maintenance. Although the flexible nature of shelterin complex impedes structural determination of the intact complex, now, all the structural domains and subcomplexes have been structurally characterized (Figure 7B). The next challenge will be to decipher the structure and dynamic regulation of the full shelterin complex to reveal how these subcomplexes, or functional units, coordinate for telomere replication and protection.
Materials and Methods
Protein expression and purification
Human TIN2TRFH (residues 2-202) (GenBank: AAF18439.1) and TPP1TBM (residues 510-544) (GenBank: AAS80318.1) fragments were cloned into modified pET-Duet vector, TIN2TRFH into promoter I, and TPP1TBM into promoter II. A 6x His tag and a 3C protease site (LEVLFQGP) were fused at its N-terminus of TIN2TRFH. The TIN2TRFH-TPP1TBM complex was coexpressed in Escherichia coli BL21 (DE3). After induction for 16 h with 0.4 mM IPTG at 25 °C, the cells were harvested by centrifugation, and the pellets were resuspended in lysis buffer (50 mM Tris-HCl, pH 8.0, 400 mM NaCl, 3 mM imidazole, 10% glycerol, 1 mM PMSF, 2 mM 2-mercaptoethanol, and homemade protease inhibitor cocktail). Cells were lysed by sonication followed by centrifugation to remove the cell debris. The supernatant was mixed with Ni-NTA agarose beads (Qiagen) and rotated for 2 h at 4 °C before elution with 300 mM imidazole. 3C protease was added at a molar ratio of 1:100 to remove the 6x His tag in the N-terminus of TIN22-202. After 3C digestion, the TIN22-202-TPP1TBM complex was further purified by gel-filtration chromatography on Hiload Superdex200 column (GE Healthcare) equilibrated with buffer (25 mM Tris-HCl, pH 8.0, 150 mM NaCl). The purified TIN2TRFH-TPP1TBM complex was concentrated to 25 mg/mL and stored at −80 °C.
Human TRF2TBM (residues 350-366) (GenBank: AAB81135.1) was expressed in E. coli BL21 (DE3) using a modified pET28b vector with a SUMO protein fused at the N-terminus after the 6x His tag. After induction for 5 h with 0.1 mM IPTG at 37 °C, the cells were harvested by centrifugation, and the pellets were resuspended in lysis buffer (50 mM Tris-HCl, pH 8.0, 400 mM NaCl, 3 mM imidazole, 10% glycerol, 1 mM PMSF, 2 mM 2-mercaptoethanol, and homemade protease inhibitor cocktail). The cells were then lysed by sonication, followed by centrifugation to remove cell debris. The supernatant was mixed with Ni-NTA agarose beads (Qiagen) and rotated for 2 h at 4 °C before elution with 250 mM imidazole. Ulp1 protease was added at a molar ratio of 1:200 to remove the His-SUMO tag. After Ulp1 digestion, the TRF2TBM peptide was further purified by gel-filtration chromatography on Hiload Superdex75 column (GE Healthcare) equilibrated with 150 mM NH4HCO3 solution. The fractions containing the peptides were collected and lyophilized.
Crystallization, data collection, and structure determination
The TIN2TRFH-TPP1TBM complex and TRF2TBM peptide were mixed at a molar ratio of 1:2 and the mixtures were used for crystallization. Crystal screening was performed with Hampton-screening kit by sitting-drop-vapor diffusion at 20 °C. The precipitant/well solutions are solution A for native sample crystals and solution B for selenomethionine (Se-Met) sample crystals (solution A: 0.2 M calcium acetate hydrate, 20% polyethylene glycol 3350, and 2 mM DTT; solution B: 0.1 M Tris-HCl, pH 8.5, 0.2 M magnesium chloride, 25% polyethylene glycol 3350, and 2 mM DTT). All crystals were gradually transferred into harvesting solutions (precipitant solution and 25% glycerol) before being flash-frozen in liquid nitrogen. SeMet-SAD diffraction data were screened and collected at the beamlines BL18U and BL19U1 of the Shanghai Synchrotron Radiation Facility with a wavelength of 0.979 Å at 100 K, and processed using HKL200049. Six selenium sites were located and refined, and the single-wavelength anomalous diffraction data phases were calculated with SHARP50. The structure refinement was done in PHENIX package51 with manual rebuilding in COOT52.
Yeast two-hybrid screening
Yeast cells growth and manipulation were done according to standard procedures. The yeast strain L40 (MATa his3Δ200 trp1-901 leu2-3112 ade2 LYS::(4lexAop-HIS3) URA3::(8lexAop-LacZ)GAL4) was used in this study. The yeast two-hybrid assays were performed with two plasmids pBTM116 (binding domain) and pACT2 (activation domain). The colonies containing both plasmids were selected on -Leu -Trp plates. The β-galactosidase activities were measured by liquid assay according to the standard manual.
Microscale thermophoresis assay
MST is a novel immobilization-free technique for the analysis of molecular interactions. MST measurements were performed with a NanoTemper Monolith NT.115 instrument (NanoTemper Technologies). In brief, in this study, 50 nM NT-650 (NanoTemper Technologies)-labeled TRF2 full-length proteins (10 μL) were first incubated for 10 min on ice with different concentrations of the TIN2FL, TIN2N, and TIN2C proteins (10 μL), respectively, in 1× PBS buffer (NaCl 137 mM, KCl 2.7 mM, Na2HPO4 10 mM, and KH2PO4 2 mM, pH 7.4). Then, 5 μL of the samples was loaded into standard treated capillaries, and MST measurements were collected at 25 °C at 30% infrared-laser power and 60% light-emitting-diode power. The laser-on and laser-off intervals were 30 and 5 s, respectively. NanoTemper Analysis 1.2.20 software was used to fit the data and to determine the apparent Kd values. All measurements were collected at least three times.
Fluorescence polarization
Different ligand proteins were diluted to a series of concentrations from 6 nM to 200 μM in 20 mM HEPES, pH 7.4, 150 mM NaCl, and 10% glycerol. The FAM-labeled molecules were used at a final concentration of 100 nM. The final volume was brought up to 100 μL with dilution buffer (20 mM HEPES, pH 7.8, 150 mM NaCl, and 10% glycerol) and incubated in the dark for 30 min. The fluorescence polarization values were measured using Synergy Neo Multi-Mode Reader (Bio-Tek) at 25 °C. Excitation wavelength was 485 nm and emission was detected at 528 nm. Fluorescence was quantitated with GEN 5 software and data were analyzed with Prism 6.
Electrophoretic mobility shift assay
The sequence of DNA template used for the assay is 5′-CTGGATCCNNNNNNNNNNNNNNNNNNNNNGTCGACAAGCTTCTCGAGAC-3′. The dsDNA was obtained by annealing this (N)21-containing template with a FAM-labeled oligo (5′-GTCTCGAGAAGCTTGTCGAC-3′), followed by filling of 5′ overhangs to form blunt ends using Klenow enzyme. Various concentrations of proteins in binding buffer (25 mM Tris-HCl, pH 8.0, 150 mM NaCl, 2 mM DTT, and 10% glycerol) were mixed with 50 nM FAM-labeled dsDNA in a total volume of 15 μL. The reaction mixtures were incubated at room temperature for 30 min before being loaded onto a 4%-20% nondenaturing polyacrylamide gel. The gels were then dried and visualized on Bio-Rad PharosFX Plus.
Size-exclusion chromatography coupled with multiangle light scattering
About 20-μL protein samples (2 mg/mL in 50 mM Tris-HCl, pH 8.0, 150 mM NaCl) were analyzed with static light scattering by injecting them into an Agilent 1260 HPLC system with a SEC column (Wyatt Technology, WTC-030S5). The chromatography system was coupled with an 18-angle light-scattering detector (DAWN HELEOS II, Wyatt Technology) and differential refractive index detector (Optilab T-rEx, Wyatt Technology). Masses (molecular weights) were calculated with ASTRA (Wyatt Technology). Bovine serum albumin (Sigma) was used as the calibration standard.
Co-immunoprecipitation
293T cells were transiently transfected by lipofectamine 3000 using 4 μg of total plasmid DNA per well in six-well dishes. Cells were lysed in IP buffer (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.05% Triton X-100, 1 mM EDTA and EGTA, 10% glycerol, 1 mM DTT, and 1 mM phenylmethylsulfonyl fluoride), and supernatants were used for immunoprecipitation with Flag-M2-beads (Sigma). Beads were washed four times and then proteins were eluted and analyzed by SDS-PAGE.
Antibodies and western analysis
The antibodies used for western blot analysis are as follows: rabbit anti-phospho-CHK1 (Cell Signaling Technology, 2348), mouse anti-phospho-CHK2 (BD Biosciences, 611570), mouse anti-γ-H2AX (Millipore, 05-636), mouse anti-TRF2 (Millipore, 05-521), rabbit anti-53BP1 (Santa Cruz, sc-22760), mouse anti-Flag (Sigma-Aldrich, F3165), mouse anti-γ-tubulin (clone GTU-488, Sigma-Aldrich, T6557), mouse anti-TRF1 (Abcam, ab10579). Rabbit anti-mTRF2 antibody was a gift from J Karlseder. Rabbit anti-mTPP1 antibody was generated using the peptide sequence CSQLLDEVREDQDHR. For immunoblotting, trypsinized cells were lysed in urea lysis buffer (8M urea, 50 mM Tris-HCl, pH 7.4, and 150 mM β-mercaptoethanol). The lysate was resolved on SDS-PAGE gel and separated proteins were then blotted on a PVDF membrane (Amersham), blocked with blocking solution (5% nonfat dry milk in PBS/0.1% Tween 20) for 1 h, and incubated with the appropriate primary antibody in blocking solution for 2 h at room temperature or overnight at 4 °C. The membranes were washed for 3 × 5 min with PBS/0.1% Tween 20 and incubated with the appropriate secondary antibody in blocking solution for 1 h at room temperature. Chemiluminescence detection was performed using an ECL Western Blotting Detection kit from GE Healthcare.
Expression vectors and shRNAs
Human and mouse WT TIN2 and mutants were cloned into pQCXIP puro retroviral expression vectors. All the constructs were confirmed by sequencing. Point mutations and shRNA-resistant sequence were introduced using side-directed mutagenesis (Stratagene). Lenti shRNA against mTIN2 (TRCN0000305996, TRCN0000305925) was purchased from Sigma. shRNA against hTIN2 was from47.
Retroviral infections
For retroviral infection, DNA constructs were transfected into 293T cells using Fugene 6 and packaged into viral particles. Viral supernatant was collected 48-72 h after transfection, filtered, and directly used to infect immortalized MEFs.
Immunofluorescence and fluorescent in situ hybridization
Cells grown on coverslips were fixed for 10 min in 2% (w/v) sucrose and 2% (v/v) paraformaldehyde at room temperature followed by PBS washes. Coverslips were blocked in 0.2% (w/v) fish gelatin and 0.5% (w/v) BSA in PBS. Cells were incubated with primary antibodies and after PBS washes, cells were incubated with appropriate Alexa Fluor secondary antibodies followed by washes in PBS + 0.1% Triton X-100. IF-FISH was carried out using a 5′-Tam-OO-(CCCTAA)4-3′ PNA telomere probe (PANAgene). DNA was stained with DAPI, and digital images were captured using Metamorph (Molecular Devices) with a Nikon Eclipse 800 microscope and an Andore CCD camera. Cells with ≥5 γ-H2AX or 53BP1-positive signals colocalizing with telomere signals are considered as TIF-positive cells53,54.
Chromosome analysis by telomere PNA-FISH and CO-FISH
Cells were treated with 0.5 μg/mL of Colcemid before harvest. Chromosomes were fixed and telomere PNA-FISH was performed with a 5′-Tam-OO-(CCCTAA)4-3′ probe (PANAgene) as described12,54. CO-FISH was used to detect both newly synthesized leading- and lagging-strand telomeric DNAs6,33. For CO-FISH, metaphase spreads were incubated sequentially with 5′-Tam-OO-(CCCTAA)4-3′ and 5′-FITC-CO-(TTAGGG)4-3′ probes. Images were captured as above. The percent of chromosome fusions observed is defined as the total number of fused chromosomes in 30-50 metaphase spreads divided by the total number of chromosomes examined × 100%. The frequency of chromatid-type and sister fusions was quantified as the number of fused chromatid (sister) ends/total number of chromatid ends × 100%.
Accession codes
Coordinate and structure factors have been deposited in the Protein Data Bank under accession codes 5XYF (TIN2TRFH-TPP1TBM-TRF2TBM).
Author Contributions
YC, SC, and ML conceived this study. CHu, JX, and YX purified the proteins. CHu and YC performed crystallization and X-ray crystallography analyses. CHu performed MST analyses. CHu, CHuang, and JL performed yeast two-hybrid assays. CHuang and JL carried out the Co-IP analyses. RR and BC carried out the IF, FISH, and CO-FISH experiments and analyzed the data. CHu, RR, CHuang, BC, and YC prepared the figures. YC, RR, SC, and ML wrote the manuscript.
Competing Financial Interests
The authors declare no competing financial interests.
Accession codes
References
Palm W, de Lange T . How shelterin protects mammalian telomeres. Annu Rev Genet 2008; 42:301–334.
de Lange T . Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev 2005; 19:2100–2110.
de Lange T . How telomeres solve the end-protection problem. Science 2009; 326:948–952.
Chen Y, Yang Y, van Overbeek M, et al. A shared docking motif in TRF1 and TRF2 used for differential recruitment of telomeric proteins. Science 2008; 319:1092–1096.
Kim H, Lee OH, Xin H, et al. TRF2 functions as a protein hub and regulates telomere maintenance by recognizing specific peptide motifs. Nat Struct Mol Biol 2009; 16:372–379.
Rai R, Hu C, Broton C, Chen Y, Lei M, Chang S . NBS1 phosphorylation status dictates repair choice of dysfunctional telomeres. Mol Cell 2017; 65:801–817.e4.
Wang F, Podell ER, Zaug AJ, et al. The POT1-TPP1 telomere complex is a telomerase processivity factor. Nature 2007; 445:506–510.
Xin H, Liu D, Wan M, et al. TPP1 is a homologue of ciliate TEBP-beta and interacts with POT1 to recruit telomerase. Nature 2007; 445:559–562.
Nandakumar J, Bell CF, Weidenfeld I, Zaug AJ, Leinwand LA, Cech TR . The TEL patch of telomere protein TPP1 mediates telomerase recruitment and processivity. Nature 2012; 492:285–289.
Denchi EL, de Lange T . Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature 2007; 448:1068–1071.
Guo X, Deng Y, Lin Y, et al. Dysfunctional telomeres activate an ATM-ATR-dependent DNA damage response to suppress tumorigenesis. EMBO J 2007; 26:4709–4719.
Wu L, Multani AS, He H, et al. Pot1 deficiency initiates DNA damage checkpoint activation and aberrant homologous recombination at telomeres. Cell 2006; 126:49–62.
He H, Multani AS, Cosme-Blanco W, et al. POT1b protects telomeres from end-to-end chromosomal fusions and aberrant homologous recombination. EMBO J 2006; 25:5180–5190.
Hockemeyer D, Daniels JP, Takai H, de Lange T . Recent expansion of the telomeric complex in rodents: two distinct POT1 proteins protect mouse telomeres. Cell 2006; 126:63–77.
Kibe T, Zimmermann M, de Lange T . TPP1 blocks an ATR-mediated resection mechanism at telomeres. Mol Cell 2016; 61:236–246.
Kim SH, Kaminker P, Campisi J . TIN2, a new regulator of telomere length in human cells. Nat Genet 1999; 23:405–412.
Ye JZ, Donigian JR, van Overbeek M, et al. TIN2 binds TRF1 and TRF2 simultaneously and stabilizes the TRF2 complex on telomeres. J Biol Chem 2004; 279:47264–47271.
Frescas D, de Lange T . Binding of TPP1 protein to TIN2 protein is required for POT1a,b protein-mediated telomere protection. J Biol Chem 2014; 289:24180–24187.
Frescas D, de Lange T . TRF2-tethered TIN2 can mediate telomere protection by TPP1/POT1. Mol Cell Biol 2014; 34:1349–1362.
Kim SH, Beausejour C, Davalos AR, Kaminker P, Heo SJ, Campisi J . TIN2 mediates functions of TRF2 at human telomeres. J Biol Chem 2004; 279:43799–43804.
O'Connor MS, Safari A, Xin H, Liu D, Songyang Z . A critical role for TPP1 and TIN2 interaction in high-order telomeric complex assembly. Proc Natl Acad Sci USA 2006; 103:11874–11879.
Takai KK, Kibe T, Donigian JR, Frescas D, de Lange T . Telomere protection by TPP1/POT1 requires tethering to TIN2. Mol Cell 2011; 44:647–659.
Frank AK, Tran DC, Qu RW, Stohr BA, Segal DJ, Xu L . The Shelterin TIN2 subunit mediates recruitment of telomerase to telomeres. PLoS Genet 2015; 11:e1005410.
Ye JZ, de Lange T . TIN2 is a tankyrase 1 PARP modulator in the TRF1 telomere length control complex. Nat Genet 2004; 36:618–623.
Zeng Z, Wang W, Yang Y, et al. Structural basis of selective ubiquitination of TRF1 by SCFFbx4. Dev Cell 2010; 18:214–225.
Liu D, Safari A, O'Connor MS, et al. PTOP interacts with POT1 and regulates its localization to telomeres. Nat Cell Biol 2004; 6:673–680.
Holm L, Rosenstrom P . Dali server: conservation mapping in 3D. Nucleic Acids Res 2010; 38:W545–W549.
Imielinski M, Berger AH, Hammerman PS, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 2012; 150:1107–1120.
Boora GK, Kanwar R, Kulkarni AA, et al. Exome-level comparison of primary well-differentiated neuroendocrine tumors and their cell lines. Cancer Genet 2015; 208:374–381.
Attwooll CL, Akpinar M, Petrini JH . The mre11 complex and the response to dysfunctional telomeres. Mol Cell Biol 2009; 29:5540–5551.
Deng Y, Guo X, Ferguson DO, Chang S . Multiple roles for MRE11 at uncapped telomeres. Nature 2009; 460:914–918.
Dimitrova N, de Lange T . Cell cycle-dependent role of MRN at dysfunctional telomeres: ATM signaling-dependent induction of nonhomologous end joining (NHEJ) in G1 and resection-mediated inhibition of NHEJ in G2. Mol Cell Biol 2009; 29:5552–5563.
Rai R, Zheng H, He H, et al. The function of classical and alternative non-homologous end-joining pathways in the fusion of dysfunctional telomeres. EMBO J 2010; 29:2598–2610.
Wang M, Wu W, Rosidi B, Zhang L, Wang H, Iliakis G . PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res 2006; 34:6170–6182.
Yan CT, Boboila C, Souza EK, et al. IgH class switching and translocations use a robust non-classical end-joining pathway. Nature 2007; 449:478–482.
Boboila C, Yan C, Wesemann DR, et al. Alternative end-joining catalyzes class switch recombination in the absence of both Ku70 and DNA ligase 4. J Exp Med 2010; 207:417–427.
Simsek D, Jasin M . Alternative end-joining is suppressed by the canonical NHEJ component Xrcc4-ligase IV during chromosomal translocation formation. Nat Struct Mol Biol 2010; 17:410–416.
Sfeir A, de Lange T . Removal of shelterin reveals the telomere end-protection problem. Science 2012; 336:593–597.
Chen Y, Rai R, Zhou ZR, et al. A conserved motif within RAP1 has diversified roles in telomere protection and regulation in different organisms. Nat Struct Mol Biol 2011; 18:213–221.
Chen C, Gu P, Wu J, et al. Structural insights into POT1-TPP1 interaction and POT1 C-terminal mutations in human cancer. Nat Commun 2017; 8:14929.
Rice C, Shastrula PK, Kossenkov AV, et al. Structural and functional analysis of the human POT1-TPP1 telomeric complex. Nat Commun 2017; 8:14928.
Shi T, Bunker RD, Mattarocci S, et al. Rif1 and Rif2 shape telomere function and architecture through multivalent Rap1 interactions. Cell 2013; 153:1340–1353.
Spengler D . The protein kinase Aurora C phosphorylates TRF2. Cell Cycle 2007; 6:2579–2580.
Fairall L, Chapman L, Moss H, de Lange T, Rhodes D . Structure of the TRFH dimerization domain of the human telomeric proteins TRF1 and TRF2. Mol Cell 2001; 8:351–361.
Broccoli D, Smogorzewska A, Chong L, de Lange T . Human telomeres contain two distinct Myb-related proteins, TRF1 and TRF2. Nat Genet 1997; 17:231–235.
Benarroch-Popivker D, Pisano S, Mendez-Bermudez A, et al. TRF2-mediated control of telomere DNA topology as a mechanism for chromosome-end protection. Mol Cell 2016; 61:274–286.
Chen LY, Zhang Y, Zhang Q, et al. Mitochondrial localization of telomeric protein TIN2 links telomere regulation to metabolic control. Mol Cell 2012; 47:839–850.
Lam YC, Akhter S, Gu P, et al. SNMIB/Apollo protects leading-strand telomeres against NHEJ-mediated repair. EMBO J 2010; 29:2230–2241.
Otwinowski Z, Minor W . Processing of X-ray Diffraction Data Collected in Oscillation Mode Methods in Enzymology. New York: Academic Press 1997:307–326.
Vonrhein C, Blanc E, Roversi P, Bricogne G . Automated structure solution with autoSHARP. Methods Mol Biol 2007; 364:215–230.
Adams PD, Grosse-Kunstleve RW, Hung LW, et al. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr 2002; 58:1948–1954.
Emsley P, Cowtan K . Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 2004; 60:2126–2132.
Takai H, Smogorzewska A, de Lange T . DNA damage foci at dysfunctional telomeres. Curr Biol 2003; 13:1549–1556.
Rai R, Chang S . Probing the telomere damage response. Methods Mol Biol 2017; 1587:133–138.
Acknowledgements
We thank staffs from BL18U1 and BL19U1 beamlines at NCPSS and Shanghai Synchrotron Radiation Facility (SSRF) for their help with crystal data collection. We are extremely grateful to National Center for Protein Sciences Shanghai (Protein Expression and Purification system, NMR system) for their instrument support and technical assistance. This work was supported by grants from the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB08010201) to ML and YC, the Ministry of Science and Technology of China (2013CB910401 to YC,2013CB910402 to ML), the National Natural Science Foundation of China (31470737 and 31670748 to YC, 31330040 and 31525007 to ML), the Basic Research Project of Shanghai Science and Technology Commission (14JC1407200 to YC), and NCI (RO1 CA129037, RO1CA202816, R21CA200506, and R21CA182280) to SC.
Author information
Authors and Affiliations
Corresponding authors
Additional information
( Supplementary information is linked to the online version of the paper on the Cell Research website.)
Supplementary information
Supplementary information, Figure S1
Mapping interaction domains between TRF2-TIN2 and TPP1-TIN2. (PDF 443 kb)
Supplementary information, Figure S2
The crystal structure of TIN2TRFH-TPP1TBM-TRF2TBM complex. (PDF 2180 kb)
Supplementary information, Figure S3
Analyses of TIN2 localization in U2OS cells. (PDF 140 kb)
Supplementary information, Figure S4
TIN2 protects telomeres from engaging in DNA damage responses. (PDF 205 kb)
Supplementary information, Figure S5
TIN2 interaction with shelterin components is required to prevent chromosome fusions. (PDF 2125 kb)
Supplementary information, Figure S6
TIN2TRFH shows distinct properties compared with TRF2TRFH. (PDF 854 kb)
Supplementary information, Figure S7
TIN2132-188 is crucial for both TRF2 and TPP1 interactions, and also maintains the correct fold of TIN2TRFH. (PDF 153 kb)
Rights and permissions
About this article

Cite this article
Hu, C., Rai, R., Huang, C. et al. Structural and functional analyses of the mammalian TIN2-TPP1-TRF2 telomeric complex. Cell Res 27, 1485–1502 (2017). https://doi.org/10.1038/cr.2017.144
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.2017.144
Keywords
This article is cited by
-
Potential effects of assisted reproductive technology on telomere length and telomerase activity in human oocytes and early embryos
Journal of Ovarian Research (2023)
-
Shaping human telomeres: from shelterin and CST complexes to telomeric chromatin organization
Nature Reviews Molecular Cell Biology (2021)
-
The role of telomere dysfunction in genomic instability and age-related diseases
Genome Instability & Disease (2021)
-
Revesz syndrome revisited
Orphanet Journal of Rare Diseases (2020)
-
Microcephalin 1/BRIT1-TRF2 interaction promotes telomere replication and repair, linking telomere dysfunction to primary microcephaly
Nature Communications (2020)
