
Abstract
Transplantation of neural progenitors derived from human embryonic stem cells (hESCs) provides a potential therapy for ischemic stroke. However, poor graft survival within the host environment has hampered the benefits and applications of cell-based therapies. The present investigation tested a preconditioning strategy to enhance hESC tolerance, thereby improving graft survival and the therapeutic potential of hESC transplantation. UC06 hESCs underwent neural induction and terminal differentiation for up to 30 days, becoming neural lineage cells, exhibiting extensive neurites and axonal projections, generating synapses and action potentials. To induce a cytoprotective phenotype, hESC-derived neurospheres were cultured at 0.1% oxygen for 12 h, dissociated and plated for terminal differentiation under 21% oxygen. Immunocytochemistry and electrophysiology demonstrated the ‘hypoxic preconditioning’ promoted neuronal differentiation. Western blotting revealed significantly upregulated oxygen-sensitive transcription factors hypoxia-inducible factor (HIF)-1α and HIF-2α, while producing a biphasic response within HIF targets, including erythropoietin, vascular endothelial growth factor and Bcl-2 family members, during hypoxia and subsequent reoxygenation. This cytoprotective phenotype resulted in a 50% increase in both total and neural precursor cell survival after either hydrogen peroxide insult or oxygen–glucose deprivation. Cellular protection was maintained for at least 5 days and corresponded to upregulation of neuroprotective proteins. These results suggest that hypoxic preconditioning could be used to improve the effectiveness of human neural precursor transplantation therapies.
Similar content being viewed by others

Main
Stroke is a devastating disease afflicting ∼700 000 people every year within the United States and causing about 150 000 deaths.1 Despite extensive research on the mechanism and treatment of stroke, very limited effective therapies are available for stroke patients. Current consent agrees that, in addition to the application of neuroprotective strategies, neuroregenerative therapies involving endogenous or exogenous approaches should be explored for repairing ischemia-damaged brain regions. In this regard, recent studies have demonstrated the potential for transplantation of neural progenitors derived from human embryonic stem cells (hESCs) to ameliorate the structural and behavioral deficits associated with cerebral ischemia animal models.2 Recent FDA clearance for the first Phase I clinical trial use of hESC transplantation therapy provides increasing expectation for hESC therapies.3
In stem cell therapy for strokes, a key step is the neural differentiation of pluripotent stem cells. Mouse ES cell can differentiate into neuronal and glial lineage cells, both in vitro and following transplantation into the ischemic brain.4, 5 Consistent with their pluripotency, neuronal and non-neuronal differentiation of human ES cells were also shown in vitro and in vivo.6 However, there have been fewer direct demonstrations of functional activities of hESC-derived neuronal cells.5, 6, 7 In the present investigation, we examined neuronal differentiation of hESCs using both morphological and electrophysiological methods.
An issue limiting the clinical application and effectiveness of stem cell therapies is the high rate of transplanted cell death. Graft survival within the ischemic core and peri-infarct regions following stroke is threatened by the release of excitotoxic neurotransmitters/factors, free radical generation and secretion of proinflammatory mediators.8 Research on grafted cells shows 30–90% cell death within a few days of transplantation.4, 9, 10 Human neural precursor transplantation studies following ischemia concur, with reports of 60–90% graft cell death.11, 12 Cell death of transplanted cells not only hampers the efficiency of the therapy, it also introduces an additional burden to the post-ischemic brain already compromised by a cellular debris load.13 Therefore, the development of novel strategies to enhance cell viability after transplantation is urgently important for more effective and efficient stem cell-based therapies.
Hypoxic or ischemic preconditioning was initially discovered as an endogenous protective mechanism in animals and later confirmed in virtually all types of cultured cells and organs.14, 15 In general, one or a series of sublethal hypoxic or ischemic insults can markedly enhance the tolerance of treated subjects to a consequent more severe insult. Hypoxic/ischemic preconditioning usually produces two phases of cytoprotection: Phase I, an acute protection that appears early and lasts for a few hours after insult and Phase II, a delayed but stronger protection which emerges many hours after hypoxia and can last for days or even weeks.14, 15 The delayed phase of hypoxic/ischemic preconditioning is mediated by synthesis of mRNAs and proteins, including the transcription factor hypoxia-inducible factor 1-α (HIF1-α) and HIF target genes, such as erythropoietin (EPO) and vascular endothelial growth factor (VEGF).14, 15 Our recent work demonstrated the prosurvival effects of hypoxic preconditioning on mouse ES cell-derived neural precursors and mesenchymal stem cells.16, 17 Neurally induced mouse embryoid bodies cultured in 1% oxygen for 8 h exhibited increased tolerance to serum deprivation in vitro and enhanced survival after transplantation to the ischemic rodent brain and heart.16, 17 More importantly, transplantation of preconditioned cells showed superior ability of improving functional recovery. Other studies have shown that hESCs cultured in low oxygen tensions comparable with the levels observed in the mammalian reproductive tract and the brain (1–5%) exert significant effects on cellular proliferation, pluripotency and maintenance of chromosomal stability.18 In fact, the physiological oxygen tension within the gray matter of the rat cerebral cortex was measured to range from 2.5 to 5.2% (19–40 mm Hg), well below standard hESC culture conditions (21% O2).19 On the basis of these findings, we proposed that under a low oxygen culture condition, hESCs should still be able to differentiate normally and meanwhile acquire enhanced tolerance to injurious insults. The increased trophic factors promoted by a sublethal hypoxia should bring out additional benefits such as stimulating neurogenesis and angiogenesis in the host tissue.
Results
hESC neurospheres and directed neural differentiation
The bone morphogenic protein (BMP) family signaling promotes embryonic stem cell self-renewal, while at the same time promotes mesodermal and trophoblast differentiation instead of neural differentiation.20, 21 hESC supplementation with BMP antagonist Noggin and bFGF produced a predominantly neuronal cell phenotype with extremely low expression of pluripotent, mesodermal and endodermal-specific genes.22 For our studies, we chose to direct hESCs to a neural phenotype using an established protocol with some modifications.22
Culture of the UCO6 hESC line on a mouse embryonic fibroblast (MEF) feeder layer allowed for efficient growth of undifferentiated but pluripotent colonies, evidenced by cellular morphology and immunostaining for pluripotent cell surface markers (Figure 1a–d). hESCs might acquire chromosomal abnormalities through enzymatic passage, particularly aneuploidy, trisomy 12 and trisomy 17.23 To prevent this, we eliminated enzymatic passaging and opted for manual dissection to better maintain chromosomal stability. Standard Giemsa banding analysis demonstrated that manual dissection prevented chromosomal abnormalities, sustaining normal cell karyotype for up to 75 passages (Figure 1e).

Neural differentiation of UCO6 hESCs. (a) Typical colony of hESCs (passage 70). (b–d) Colonies express pluripotent markers TRA-1-60 (b) and SSEA4 (c), but are negative for SSEA-1 (d). (e) Normal karyotypic analysis of passage 75 UCO6 colonies. (f) Twenty-eight day floating neurosphere (∼200 μm in size). (g–j) After beginning terminal differentiation, cells exhibit increasingly neural morphology after 1 day (g), 7 days (h) and 14 days (i). Cells exhibit significant neuritic extensions and dendritic spine formation (white arrows) after 21 days terminal differentiation (j)
To induce neural differentiation, manually isolated colonies were cultured as floating neurospheres for 42 days (Figure 1f). After plating for adhesion, polarized individual cells migrated outward from the spherical center after 24 h (Figure 1g). After 7 and 14 days, cell body size and projection length increased; by 21 days cellular projections increased not only in size but also in density. Individual cells displayed multiple neurite outgrowths and dendritic spines, characteristics typical of an immature neuronal phenotype (Figure 1h–j).
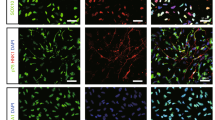
To verify that the differentiating cells were neural in nature, immunostaining was performed at various stages of development. Twenty-four hours after plating, the majority of cells were positive for the neural precursor protein nestin (87.2±1.97%). Importantly, cells positive for the pluripotent cell surface antigen stage-specific embryonic antigen 4 (SSEA-4) were virtually non-existent (Figure 2a). Beginning on day 3 of terminal differentiation, expression of the medium length neurofilament polypeptide (NF-M), a neuronal-specific protein marker expressed by mature neurons, can be observed in a portion of differentiating cells (Figure 2b). Neuronal maturation continued and by 7 days, the neuronal protein markers βIII-tubulin and NeuN were expressed in 76.98±5.7 and 48.19±1.8% of total cells, respectively, indicating the formation of a neuronal cell population (Figure 2c). Allowing cells to differentiate for longer time periods lead to increased neural maturation. After 21 days, 72.7±4.2% of cells were positive for NF-M (Figure 2f and g). Also, many broad axonal structures were positive for both NF-M and the neuronal-specific protein microtubule-associated protein-2 (MAP2; Figure 2f and g). Twenty-eight days after beginning terminal differentiation, some axonal projections expressed myelin-binding protein (MBP), suggesting axonal myelination and sheath formation (Figure 2h). Glial cells positive to glial fibrillary acidic protein (GFAP) and the oligodendrocyte marker O4 were observed after 7-day differentiation. GFAP was expressed in 19.2±2.4% of all cells, while O4-positive cells were rarely observed (Figure 2d and e). Glial proliferation continued after a month differentiation, likely providing enhanced trophic supports for maturing neurons. To some extent, this mixed culture provides an environment mimicking in vivo cell compositions (Figure 2i).

Neural gene expression in differentiating hESC-derived neural progenitor cells. (a) Cells exhibit high expression of the neural precursor marker nestin (green) and very low expression of SSEA-4 (red) 1 day after cell adhesion. (b and c) Three days into terminal differentiation, nestin (green) positive, elongated cells begin to express medium chain neurofilament (NF-M; red). (d) Seven days after plating, the neuronal markers βIII-tubulin (green) and NeuN (red) were highly expressed. (e) Expression of the astrocyte marker GFAP is observed after 7 days. (f) Very few O4-positive immunoreactive cells were found in these cells. (g and h) Extensive NF-M (green) and microtubule-associate protein-2 (MAP2; red) immunoreactive projections were present in cells of at 21 days into differentiation. (i) Twenty eight days after plating, some projections are positive for myelin-binding protein (MBP), indicating axonal myelination. (j) Extensive beds of GFAP-positive astrocytes can also be observed after 28 days into terminal differentiation. All blue staining indicates Hoechst-positive nuclei
Functional expression of voltage- and ligand-gated channels in hESC-derived neurons
To verify that neuron-like cells derived from hESC-derived neurospheres were physiologically active, patch clamp whole-cell recording was performed on cells at various stages of maturation. In cells undergoing 21 days terminal differentiation, stepwise depolarizing pulses from a holding potential of −70 mV evoked a rapidly activating/inactivating inward current and a steady outward current of increasing amplitudes (Figure 3a). Addition of 500 nM tetrodotoxin (TTX) to the extracellular solution abolished the inward current, corroborating the expression of TTX-sensitive sodium channels, INa, in these cells (Figure 3b).

Neuronal channel and synaptic activities in hESC-derived neurons. (a) Depolarization steps from a −70 mV holding potential induced rapidly activating inward currents followed by slow inactivated outward currents. (b) Administration of 500 nM TTX completely blocked the inward current, confirming its Na+ current nature. (c) The current–voltage relationship of Na+ currents in 7- (closed triangles) and 21-day (open triangles) differentiating cells. (d) In the presence of TTX, most cells showed fast- and non-inactivated outward currents. The fast inactivating IA current is outlined by a rectangular box, while the approximate start of steady state IK is labeled with an arrow. (e) The application of 10 mM TEA attenuated both outward currents, consistent with the sensitivity of K+ channels to TEA. (f) The I–V relationship of the IK current in hESC-derived neuron-like cells 7 days (closed triangles) and 21 days (open triangles) in differentiation. (g and h) Block of the fast outward currents by 4-aminopyridine, suggesting the existence of the IA current in these cells. (i) Summary graph of peak current of INa, IK and IA. *P<0.05 between 7 and 21 days; #P<0.05 between 14 and 21 days. (j) After 28-day terminal differentiation, expression of NMDA receptors was indicated by immunostaining of the NR1 subunit in hESC-derived cells. (k) Extensive expression of the synaptic vesicle protein synaptophysin can be observed overlaying with NF-M after 28 days in differentiation. (l) Higher magnification image shows punctate expression of synaptophysin in differentiated cells. (m) Patch clamp whole-cell recordings on cells of 28-day neural differentiation revealed functional NMDA (left) and AMPA (right) currents. (n) Representative trace shows spontaneous miniature potentials over a 2 min recording in cell after 35-day differentiation
In the presence of TTX, membrane depolarization triggered sustained outward currents in most cells, containing both a fast inactivating (outlined by rectangular box) and a slow inactivating outward current (Figure 3c). The broad spectrum potassium channel blocker tetraethylammonium bromide (TEA; 10 mM) inhibited both fast- and slow-inactivating outward currents, indicating that these were TEA-sensitive K+ currents (Figure 3d). Addition of 4-aminopyridine (5 mM) selectively blocked the fast-inactivating current (Figure 3e and f), further suggesting the existence of the A-type K+ current, IA, in these cells. Using current–voltage plots of both INa and IK, determination of half-maximal currents indicated that both these two currents showed typical voltage-dependent activations (Figure 3g and h). All three currents showed time-dependent increases during neuronal maturation (Figure 3i).
Immunohistochemical staining and whole-cell recording were applied to detect glutamate receptors and synaptogenesis in differentiating cells (Figure 3j–l). The glutamate receptor subtype-specific agonist N-methyl-D-aspartic acid (NMDA) or α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) was locally applied to hESC-derived cells under whole-cell voltage clamp. Administration of 200 μM NMDA and 10 μM glycine elicited an inward current in 43% of cells 28 days into differentiation, with peak current of 843±329 pA and steady state current of 698±294 pA (n=14, Figure 3m). Similarly, 45% of these cells exhibited a fast inactivating inward current (peak current=201±30 pA) in response to application of 100 μM AMPA (n=11, Figure 3m).
Miniature postsynaptic currents (mPSCs), measures of spontaneous release and subsequent uptake of neurotransmitters at the synaptic cleft, were recorded in hESC-derived cells. After 35 days into terminal differentiation, most cells of neuronal morphology exhibited an extensive number of spontaneous inward events (Figure 3n). A comparison of 28- and 35-day measurements revealed an increase in both the frequency of events (0.85±0.10 versus 2.22±0.35) and the mean peak amplitude (21.32±0.14 versus 29.92±0.34) over time.
Hypoxic preconditioning increases the tolerance of hESC-derived cells
To induce a cytoprotective phenotype of hESC-derived neural cells, neurospheres were cultured for 41 days at 21% oxygen and then placed in 0.1% oxygen for different hours. The hypoxia procedure up to 72 h did not cause significant cell death measured by Trypan blue uptake in gently dissociated neurospheres immediately after hypoxia. Cultures of neural precursors in 0.1% oxygen for 12 h showed a reduction in spontaneous cell death of Trypan blue-positive cells compared with 21% oxygen maintained controls (21.6±3.0 versus 41.4±1.8%; n=4 separate experiments). This early protective effect was transient, disappearing after 24-h hypoxic culture when basal cell death between hypoxic and normoxic groups was comparable (32.4±2.5 and 41.4±1.8%; n=4). We therefore determined that a 12-h exposure to 0.1% oxygen induced a rapid phase of preconditioning.
We next determined whether a prolonged, delayed phase of preconditioning could be initiated in human neural cells. To induce delayed Phase II preconditioning, cells received an initial hypoxic exposure, followed by return to 21% oxygen for up to 5 days. Cells were then challenged with a lethal insult common to cerebral ischemia: hydrogen peroxide (H2O2; 500 μM)) or glucose and oxygen deprivation (OGD). Control hESC-derived neural cells exhibited high expression of nestin and few terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end labeling (TUNEL)-positive nuclei (Figure 4a). After 24-h H2O2 treatment, non-preconditioned cells showed extensive numbers of TUNEL-positive nuclei, whereas cells that received preconditioning were more resistant to H2O2-induced death and maintained membrane/nuclear integrity (Figure 4b and c). Quantification of TUNEL-positive cells confirmed increased survival of preconditioned neural cells compared with non-preconditioned cells 3 and 5 days after reoxygenation (Figure 4d). Preconditioned cells were also more tolerant to OGD than non-preconditioned cells, displaying a ∼50% reduction in total and neural precursor cell death 3 and 5 days after reoxygenation (Figure 4e).

Hypoxic preconditioning enhances cellular tolerance to lethal insults. (a) Neural precursors stained with Hoechst (blue) and nesting (red), there are very few TUNEL-positive (green) cells, suggesting high viability under our control condition. (b and c) The insult of hydrogen peroxide (H2O2, 500 μM) induced significantly cell death in cultures with normoxic condition (b), exhibited as increased TUNEL-positive cells. On the other hand, hypoxic-preconditioned cells showed high resistance to the same insult (c). (d and e) TUNEL-positive cells induced by H2O2 or oxygen glucose deprivation and their co-labeling with Hoechst (all cells) or nestin (neural progenitors) at different days after termination of the 12-h hypoxic pretreatment. Total cell death (TUNEL/Hoechst colocalization) and neural precursor cell death (TUNEL/nestin colocalization) were significantly reduced in preconditioned cultures after H2O2 treatment (d) or oxygen glucose deprivation (e). *P<0.05 versus normoxic culture
Previous studies have shown that culture conditions below 5% oxygen can enhance proliferation of neural and embryonic stem cells.24, 25 Increased cellular proliferation after hypoxia could partly account for increased cell counts. To evaluate this possibility, cells were pulsed overnight with bromodeoxyuridine (BrdU) that can incorporate into newly synthesized DNA of proliferating neural cell types.26 Immunocytochemical analysis of BrdU uptake after 3 days showed that both normoxic and hypoxic nestin-positive cells were highly proliferative (46.8±9.4 versus 53.2±7.9, respectively). No statistical difference was observed between these two groups (P=0.41). Immunostaining also showed very little proliferation of non-nestin-positive cells in both groups (<1% of total cells), further confirming the formation of a neurogenic, non-pluripotent cell population under our experimental condition.
Time course experiments showed no cellular protection within 24 h after reoxygenation in hypoxia-treated cells. This observation was consistent with the notion that Phase II protection requires a time-dependent process of gene regulation.14, 15 To verify this, protein and mRNA levels of cytoprotective and cell death associated genes after preconditioning were compared with normoxic controls. RT-PCR analysis showed that HIF1-α mRNA levels were increased after only 3-h culture at 0.1%, with much greater mRNA after 12- and 24-h hypoxic exposure (Figure 5a). The mRNA expression of the Bcl-2 family gene BNIP3, an HIF1-α target linked to delayed neuronal death through stabilization of p53 tumor suppressor during chronic hypoxia, was reduced (Figure 5a). On the other hand, elevated HIF1-α mRNA levels were maintained for at least 24 h after returning to 21% oxygen (Figure 5a). At the protein level, we observed HIF1-α and HIF2-α stabilization and upregulation after 12-h hypoxic culture (Figure 5b). Increased activation of the MAP kinase Akt by phosphorylation at Ser 473 was also observed by 3-h hypoxic culture and remained elevated on reoxygenation, along with increases in hypoxia-responsive proteins EPO and Bcl-2 (Figure 5c). Protein upregulation appeared later for EPO, EPO receptor (EPOR), Bcl-2 and VEGF (Figure 5c).

Hypoxic preconditioning induced gene regulation. (a) The HIF1-α mRNA level in hESC neurospheres was upregulated after 3–24 h under the 0.1% oxygen condition, revealed by RT-PCR measurements. The last column shows that the HIF1-α level remained high after finishing a 12-h hypoxia plus 12-h back to 21% oxygen (reoxygenation). No change in the expression of the proapoptotic gene BNIP3 was observed. β-Actin was used as a loading control. (b) Western blot analysis shows the upregulation of the protein levels of HIF1-α and pAkt induced by the hypoxia treatment. The Akt activation (phosphorylation of S473) was inhibited by the PI3K inhibitor Wortmannin that also partially inhibited hypoxic stabilization of HIF1-α. (c) Western blot analysis showed significant upregulation of several survival genes, including HIF1-α, HIF2-α, pAkt, EPO and Bcl-2 after the 12-h 0.1% oxygen treatment. α-Tubulin was used as a loading control. (d) Gene regulation after the termination of hypoxic treatment. Different time points after hypoxia, cells were returned to 21% oxygen. The protective gene bcl-2 decreased and proapoptotic gene Bax increased after reoxygenation. However, a significant upregulation of EPO, mature VEGF and Bcl-2, as well as a downregulation of Bax, were evident after 72-h reoxygenation. *P<0.05 between normoxic controls and 12-h hypoxic culture; **P<0.05 between normoxic controls and 72-h reoxygenation
Consistent with the lack of protection 24 h after returning to 21% oxygen, the gene expression pattern displayed a shift away from cytoprotection at this time, as evidenced by the downregulation of EPO and Bcl-2 levels concurrent with upregulation of Bax (Figure 5d). However, at 72-h time point a subsequent re-elevation of measured cytoprotective proteins (EPO, 42 kDa-VEGF, Bcl-2) and decline in apoptotic factors such as Bax was evident (Figure 5d). Seventy two hours after return to 21% oxygen, EPO and VEGF expression was increased to 58.5±0.11 and 39.6±0.12%, respectively (P<0.05, n=3), whereas the Bcl-2/Bax ratio increased 44.2±0.14% compared with non-preconditioned levels (P<0.05, n=3). These findings show a biphasic protein response after preconditioning directly corresponding to observed cytoprotection.
Whether HIF1-α stabilization is dependent on phosphoinositide 3-kinase (PI3K)/Akt signaling is a matter of question.27, 28 During this study, we observed a significant upregulation of phospho-Akt before HIF1-α stabilization (Figure 5b). To test whether Akt activation regulates HIF stabilization, the PI3K inhibitor Wortmannin (1 μM) was added to cells concurrent with the initiation of 12-h hypoxic culture. Protein analysis revealed that PI3K inhibition by Wortmannin completely attenuated Akt phosphorylation and reduced HIF1-α protein levels by ∼23% (Figure 5b).
Preconditioning increases the neurogenic capacity of human neural precursors
Finally, we wanted to ascertain whether the hypoxic preconditioning affected the neurogenic or functional potential of hESC-derived progenitor cells. To test this, cells were given the preconditioning stimulus, plated and analyzed after 7 days of neural differentiation by immunocytochemistry (Figure 6a). Preconditioned cells exhibited a significantly higher percentage of NeuN-positive cells compared with non-preconditioned controls (58.65±5.4 versus 48.19±1.8; P<0.05; Figure 6b). No significant differences in GFAP-positive cells were observed between normoxic and preconditioned groups (Figure 6b).

Hypoxic preconditioning increases hESC neuronal differentiation. (a) Immunocytochemistry of normoxic (left panel) and preconditioned (right panel) cells 7 days into terminal differentiation. Both showed high expression of neuronal and astrocyte markers NeuN (green) and GFAP (red). Hoechst (blue) indicated total cells. (b) Quantification of neuronal (NeuN/Hoechst colocalization) and glial (GFAP/Hoechst colocalization) lineage cells after 7 days differentiation. The bar graph shows preconditioning (gray bars) increased neuronal yield (NeuN-positive cells) compared with normoxic controls (blue bars). (c) Representative action potential traces evoked by injection of 30 pA current show functional activity of derived neurons over a 21-day period. (d and e) The amplitude and number of evoked action potentials significantly increased from 7 to 21 days in both normoxic (blue bars) and preconditioned cells (gray bars). Preconditioned cells showed a trend of increased size and number of action potentials. (f) Action potential half-width was significantly reduced in both normoxic and preconditioned cells. Preconditioned cells showed a trend of sharper action potentials. * and **P<0.05 compared with 7-day cells
To determine the effects of hypoxic preconditioning on generation of action potentials, cells of 7 and 21 days in differentiation were analyzed using current clamp recordings. Compared with 7-day cells, evoked spikes of 21-day cells showed increasing amplitude, decreasing width and greater numbers of events per depolarization (Figure 6c). Statistical analyses revealed no significant difference in amplitude, frequency or half-width of action potentials between preconditioned and non-preconditioned cells (Figure 6d–f).
Discussion
Cell transplantation therapy using pluripotent embryonic stem cells is an imperative component of the potential regenerative medicine. To fulfill the goal of cell replacement after ischemic stroke, cell survival and directed neural differentiation to specific functional neural cell types after engraftment are two key steps. Although priming cells before transplantation has received increased attention,16, 17, 29 harnessing this phenomenon for use in human neural precursors has not been explored. To the best of our knowledge, this is the first report of the effects of hypoxic preconditioning on hESC-derived neural precursors. We show in this present investigation that the preconditioning strategy with sublethal hypoxia is an effective way of promoting survival of human ES cell-derived neural progenitors. Previous research has shown neuronal differentiation and maturation of mouse or human ES cells in vitro and after transplantation,6 but little evidence is available so far to demonstrate functional neuronal activities, such as action potential generation and spontaneous neurotransmitter release in neurally differentiating hESCs. Our investigation provides evidence that neuronal cells derived from hESCs not only develop neuronal morphology, but also exhibit membrane excitability typical of glutamatergic neurons.
The present investigation shows that brief exposure of human neural precursor cells to hypoxic conditions can significantly enhance cellular tolerance to consequent lethal insults. This cytoprotective phenotype is maintained for at least 5 days after the hypoxic stimulus, thereby promoting cell survival during the critical acute phase after transplantation, during which 30–90% of transplanted cells succumb to environmental stresses within the host milieu.4, 11, 12 We also show that hypoxic preconditioning does not increase neural precursor proliferation or detrimentally affect neuronal differentiation. Both control and preconditioned hESC-derived cells exhibit functional maturation of voltage and ligand-gated currents as well as action potential generation.
In ES cells, HIF2-α increases Oct-4 expression, reduces cell differentiation and may promote tumor formation.30 Although our results found upregulation of hypoxia-regulated genes, including HIF α-subunits and pAkt, we did not observe enhanced proliferation after preconditioning. This could be due to the brief hypoxic exposure used (12 h in our study versus 72 h in above investigations). Supporting this idea, it was shown that mRNA expression of the mitogen FGF-8 in neural stem cells was not increased until 4 days of hypoxic culture.25 VEGF signaling through HIF1-α was previously demonstrated as a requirement for neural precursor proliferation.31 In the present investigation, VEGF was not upregulated until cells were returned to 21% oxygen and, importantly, Oct-4 expression was unchanged during and after preconditioning. These findings suggest the mild hypoxic treatment for priming hESCs did not trigger a cascade leading to increased cellular proliferation.
Preconditioned cells exhibited a significantly higher percentage of NeuN-positive cells compared with normoxic maintained controls. It is possible that enhanced neuronal formation could be due to upregulation of hypoxia-responsive genes, including tyrosine hydroxylase (TH).32 Hypoxic culture was shown to increase both the number of TH-positive neurons and dopamine production from isolated rat mesencephalic precursors.25 Neuronal formation from telencephalic precursors also increased in 3% oxygen, but neuronal subtype changed from GABAergic to glutamatergic.24 Additional data suggest that both the antiapoptotic gene Bcl-2 and the angiogenic factors EPO and VEGF can enhance neuronal protein expression and neurogenesis.33, 34 These findings suggest that preconditioning may affect both the neurogenic potential and neuronal subtype specification from neural precursors.
The necessity of HIF stabilization and downstream effectors during hypoxia and preconditioning has been well established.35 Our data showed that hypoxia stabilized HIF1-α, HIF2-α and Akt activation, followed by delayed upregulation of EPO, VEGF and Bcl-2. VEGF activation and Akt phosphorylation were required for neuronal survival after preconditioning.36 Recent work has begun to discern the importance between HIF1-α and HIF2-α subunit expression and target gene activation during neuronal cell death. HIF2-α was shown to drive astrocytic expression of EPO and increase neuronal survival, whereas HIF1-α regulated VEGF and LDH expression.37 Similarly, conditional HIF1-α deletion caused increased HIF2-α expression and EPO upregulation, suggesting potential overlap in α-subunit function.38 Future hypoxic preconditioning studies on human neural precursor survival should examine whether HIF1-α and/or HIF2-α stabilization are required for enhanced cell survival.
In summary, we have demonstrated the directed neural differentiation of the UCO6 hESC line and characterized the functional maturation of hESC-derived neurons. Furthermore, we have shown that brief exposure of human neural precursors to sublethal hypoxia can attenuate cell death induced by injurious events common to ischemic stress. Preconditioning had no detrimental effect on neural differentiation, neuronal function or proliferation. HIF stabilization and activation of several key neuroprotective genes most likely contribute to the cytoprotection. Transplanting preconditioned cells may also improve endogenous neurogenic and angiogenic responses within damaged tissue.39 It is thus suggested that hypoxic preconditioning represents a clinically applicable regime to improve ES cell transplantation therapies.
Materials and Methods
Human ES cell cultures
The UC06 human ES cell line (passages 45–70; University of California at San Francisco) was expanded on a feeder layer of mitotically inactivated MEFs in a growth medium of DMEM, 20% knockout serum replacement (Invitrogen, Carlsbad, CA, USA), 2 mM L-glutamine, 100 μM β-mercaptoethanol, 1% non-essential amino acids and 10 ng/ml recombinant human FGF-2 (R&D Systems, Minneapolis, MN, USA). To maintain pluripotency, colonies were manually separated from feeder cells and morphologically identifiable differentiated colonies. Passaging was performed every 3–5 days onto 60 mm dishes using a 20-gauge needle. The undifferentiated status of UCO6 colonies was confirmed through routine immunostaining for multiple pluripotent markers, including Oct-4, SSEA4, tumor rejection antigen 1-60 (Tra-1-60) and Tra-1-81. The karyotype of undifferentiated cells was determined every 10 passages to verify the absence of chromosomal abnormalities. All research using hESCs adhered to the guidelines established by the National Academy of Sciences.
Neural differentiation of the UCO6 hESC line
Neural differentiation from manually isolated pluripotent UC06 colonies was achieved as previously described by Itsykson et al.22 Briefly, undifferentiated hESC colonies were cultured as floating aggregates for 42 days in ultra low-binding 60 mm dishes (Corning, Corning, NY, USA). The aggregates were maintained in neural precursor media consisting of DMEM/F12, 1 × B27 (Invitrogen), 2 mM glutamine, 1 × penicillin/streptomycin and 20 ng/ml FGF-2. To inhibit non-neural differentiation, aggregates were also supplemented during the initial 21 days with 500 ng/ml recombinant mouse Noggin (R&D Systems). Media were changed every 3 days, during which aggregates over 200 μm in diameter were dissociated. After 42 days, aggregates were partially dissociated through gentle trituration and plated on poly-D-lysine (0.5 mg/ml; MP Biomedical, Solon, OH, USA)/laminin (20 μg/ml; BD Biosciences, San Jose, CA, USA) coated coverslips or chamber slides for terminal differentiation. Terminal differentiation was achieved by allowing plated cells to mature for up to 42 days without FGF-2 or Noggin supplementation.
Immunocytochemistry
Cells were fixed with 4% paraformaldehyde (PFA) for 15 min and then permeabilized with 1% Triton X-100 for 10 min. Coverslips were then incubated in 5% normal horse serum (Invitrogen) for 1 h before incubation with primary antibodies overnight at 4°C: 145 kDa neurofilament (MAB1621; 1 : 500), MAP-2 (AB5622; 1 : 500), βIII-tubulin (MAB1637; 1 : 500), synaptophysin (MAB5258; 1 : 1000), human nuclei (MAB1281; 1 : 100), NeuN (MAB377; 1 : 500), O4 (MAB345; 1 : 25), MBP (AB980; 1 : 1000), GFAP (AB1540; 1 : 1000; Chemicon, Billerica, MA, USA); NMDAR1 (05-432; 1 : 250; Upstate/Millipore, Billerica, MA, USA); human nestin (MAB1259; 1 : 500; R&D Systems). Following primary antibody incubation, cells were washed with 1 × PBS, incubated with secondary antibody Alexa Fluor 488-conjugated anti-rabbit or anti-mouse IgG, Cy3-conjugated anti-mouse IgG, or Cy5-conjugated goat anti-rat IgG (Molecular Probes, Invitrogen), washed and mounted with ProLong AntiFade (Invitrogen). Staining was visualized by fluorescent microscopy (BX61; Olympus, Tokyo, Japan).
For analysis of proliferation, cells were incubated overnight with the thymidine analog BrdU (Sigma-Aldrich, St. Louis, MO, USA; 10 μg/ml). Coverslips were then washed with fresh neural precursor medium, fixed in 4% PFA and immunostained for human nestin. To quantify BrdU incorporation, cells were again fixed in 4% PFA, washed, methanol fixed for 14 min and allowed to air-dry. After rehydrating with PBS, coverslips were incubated with 2 N HCl for 1 h at 37°C. Sections were treated with 0.1 M borate buffer at pH 8.4 for 10 min, followed by 45 min in 0.2% Triton X-100. After blocking in 5% horse serum, cells were incubated with rat anti-BrdU (1 : 500, Abcam, Cambridge, MA, USA) primary antibody overnight at 4°C followed by rinsing in PBS before incubation with Cy5 goat anti-rat IgG (1 : 500, Molecular Probes, Invitrogen) secondary antibody for 1 h at room temperature. Coverslips were mounted with ProLong Antifade (Invitrogen) for visualization.
Whole-cell patch clamp recordings
Conventional whole-cell recordings were performed in dissociated cells to record voltage-gated Na+ and K+ currents, spontaneous mPSCs and ligand-gated NMDA or AMPA evoked currents. Briefly, coverslips containing neural-induced hESCs were placed on the stage of an inverted microscope for patch clamp recordings using an EPC-9 amplifier (HEKA, Lambrecht, Germany). Recording electrodes of 6–15 MΩ were pulled from Corning Kovar Sealing #7052 glass pipettes by a Flaming-Brown micropipette puller (Corning, NY, USA). The offset potential of the tip was routinely adjusted after immersion into recording solution. All single measure receptor currents were recorded at −70 mV holding potentials. Recordings of mPSCs were performed at −60 mV holding potential. Currents and traces were acquired through the PULSE/PULSEFIT program and filtered at 3 kHz by a 3-pole Bessel filter (HEKA). Measurement of ligand-gated channel currents was achieved through use of the VM8 voltage valve control drug delivery system (ALA Scientific Instruments, Westbury, NY, USA). Recordings were performed at room temperature and pH ∼7.3. Recording solutions used contained (in mM): NaCl 115, MnCl2 2, TEA 10, HEPES 10, glucose 10 (external solution for inward Na+ current); CsCl 130, NaCl 5, HEPES 10, BAPTA 1, TEA 6 (internal solution); NaCl 115, KCL 2.5, MnCl2 2, HEPES 10, BAPTA 0.1, glucose 10, TTX 0.1 μM (external solution for outward K+ current, mPSCs); KCl 120, MgCl2 1.5, Na2-ATP 2, CaCl2 1, BAPTA 1, HEPES 10 (internal solution); NaCl 120, KCl 3, CaCl2 2, HEPES 10, glucose 10, TTX 0.5 μM (external solution for NMDA, AMPA currents); CsCl 120 Na2-ATP 2, BAPTA 0.5, HEPES 10.
Hypoxic preconditioning
Human ESCs in 100-mm dishes underwent neural induction for 42 days before transplantation or in vitro terminal differentiation. For hypoxic preconditioning treatment, hESC aggregates of 41 days differentiation were placed in a humidified chamber with 0.1–0.3% oxygen, 5% CO2 and balanced nitrogen at 37°C. Trypan blue exclusion assay showed that the low O2 treatment for up to 12 h did not cause cell death. After 12-h hypoxic exposure, dishes were removed and aggregates triturated and harvested for transplantation or plated on 35-mm coverslips or chamber slides for in vitro experimentation.
TUNEL staining
Both Trypan blue staining and TUNEL staining have been used for detecting general cell death disregarding the death mechanism (apoptosis and necrosis).17, 40 TUNEL staining, however, has the advantage of double staining with another marker to reveal specific cell types of affected cells. TUNEL staining was performed according to the manufacturer's protocols (Promega, Madison, WI, USA). Cells maintained on 12-mm coverslips were fixed with 4% PFA, followed by permeabilization with ethanol: acetic acid and 1% Triton X-100. Cells were then incubated in equilibration buffer, followed by TdT enzyme incubation. After confirmation of positive TUNEL staining, cells were further immunostained for human nestin and Hoechst. Total cell death was estimated using ImageJ software (NIH, Washington, DC, USA) through comparison of TUNEL/Hoechst colocalization versus all Hoechst-positive cells. To measure neural precursor cell death, colocalization of TUNEL/nestin/Hoechst-positive cells was compared with all nestin/Hoechst co-labeled cells.
Hydrogen peroxide and OGD insult
Non-preconditioned hESCs were subjected to varying concentrations of H2O2 (0–10 mM) to determine control cell death. Cytoprotective effects of preconditioning were determined as follows: 12 h 0.1% oxygen culture followed by 24–120 h reoxygenation to allow upregulation of cytoprotective compounds. After hypoxic culture and reoxygenation, preconditioned hESCs and non-preconditioned hESCs were treated with H2O2 for 24 h. Cells were then fixed by 4% PFA and TUNEL staining colocalized with Hoechst and nestin used to determine total cell death and neural precursor cell death. To test for OGD sensitivity, non-preconditioned hESCs and preconditioned hESCs were gently washed twice in glucose-free DMEM/F12, followed by culture in 95% N2/5% CO2 and DMEM/F12 without glucose (US Biological, Swampscott, MA, USA) or B27 for 4 h after 24–120 h reoxygenation of HP human ESCs. Cell death was determined through TUNEL staining colocalization with Hoechst and nestin.
Reverse transcriptase-polymerase chain reaction
RNA was isolated from cell aggregates by the RNeasy RNA extraction kit (Qiagen, Valencia, CA, USA). Complementary cDNA was formed from 0.250 to 1 μg RNA using Advantage RT for PCR protocol (Clontech, Mountain View, CA, USA). PCR reactions using standard protocols with Taq DNA polymerase were performed on a Px2 Thermocycler (Thermo Scientific, Waltham, MA, USA). The following conditions were used: 94°C 2 min 45 s, 50–60°C 45 s, 72°C 10 min, 25–30 cycles. PCR products were visualized on a 2% agarose gel. The following primer sequences were used: HIF1-α forward (F) – ctgaccctgcactcaatcaa, reverse (R) – tccatcggaaggactaggtg; BNIP3 F – agggctcctgggtagaactg, R – actccgtccagactcatgct; β-actin F – agaaaatctggcaccacacc, R – ggggtgttgaaggtctcaaa.
Immunoblotting
Cells were collected by centrifugation or cell scraping, and protein isolated with RIPA buffer containing protease inhibitor cocktail (Sigma-Aldrich). Protein concentrations were determined using bicinchoninic acid assay (BCA; Pierce, Rockford, IL, USA). Proteins were then separated by SDS-page gel electrophoresis, followed by transfer to PVDF membrane (Biorad, Hercules, CA, USA). Membranes were blocked for 1 h with 5% BSA containing phosphatase inhibitors (50 mM NaF, 200 μM Na3VO4) before overnight incubation with primary antibodies: VEGF (sc-507; 1 : 1000), Flk-1 (sc-6251; 1 : 1000), EPOR (sc-697; 1 : 1000), Akt (sc-8312; 1 : 1000), Bcl-2 (sc-7382; 1 : 500), Bcl-Xl (sc-8392; 1 : 500), BDNF (sc-546; 1 : 1000), α-tubulin (sc-5286; 1 : 5000), EPAS-1 (sc-28706; 1 : 2000), EPO (sc-7956; 1 : 2000; Santa Cruz Biotechnology, Santa Cruz, CA, USA), phospho-Akt (S473) (#9271; 1 : 1000), NF-ÊB (#3035; 1 : 2000; Cell Signaling Technology, Danvers, MA, USA), GLUT-1 (AB1340; 1 : 7500), 17 kDa Caspase 3 (AB3623; 1 : 2000), FGF-2 (AB1458; 1 : 500; Chemicon/Millipore), HIF1-α (NB100-479; 1 : 2000; Novus Biologicals, Littleton, CO, USA), Bax (no. 54179; 1 : 500; Anaspec, Fremont, CA, USA). Blots were washed, antigen binding detected by incubation with anti-rabbit or mouse HRP-conjugated secondary antibodies (Biorad) and detection by ECL method (Thermo Scientific). Chemiluminescence was detected on CL-XPosure radiographic films (Thermo Scientific) and intensity analyzed by ImageJ software (NIH). To compare relative protein intensity across groups, loading was normalized to either β-actin or α-tubulin expression before analysis.
Cell counting
Randomly selected fields from 12 mm coverslips were imaged at × 20 and × 40 magnification from at least three separate experiments. Approximately five visual fields were obtained per coverslip. The expression levels of proteins are based on Hoechst staining for total cell population. Multinucleated clumps of cells were excluded from cell counting analysis.
Statistical analysis
Data were graphed and analyzed using either Prism (GraphPad Software, San Diego, CA, USA) or SigmaPlot (SyStat Software, San Jose, CA, USA) software. Student's two-tailed t-test was used for comparison of two experimental groups. Multiple comparisons were performed using one-way ANOVA followed by Tukey post-test. Significance was determined based on standard error of mean and defined as P<0.05.
Conflict of interest
The authors declare no conflict of interest.
Abbreviations
- hESCs:
-
human embryonic stem cells
- HIF1-α:
-
hypoxia-inducible factor-1-α
- EPO:
-
erythropoietin
- VEGF:
-
vascular endothelial growth factor
- BMP:
-
bone morphogenic protein
- NF-M:
-
medium length neurofilament polypeptide
- MAP2:
-
microtubule-associated protein-2
- MBP:
-
myelin-binding protein
- GFAP:
-
glial fibrillary acidic protein
- TTX:
-
tetrodotoxin
- TEA:
-
tetraethylammonium bromide
- 4-AP:
-
4-aminopyridine
- H2O2:
-
hydrogen peroxide
- OGD:
-
oxygen glucose deprivation
- EPOR:
-
EPO receptor
- PI3K:
-
phosphoinositide 3-kinase
- TH:
-
tyrosine hydroxylase
- MEFs:
-
mouse embryonic fibroblasts
- SSEA4:
-
stage-specific embryonic antigen 4
- Tra-1-60:
-
tumor rejection antigen 1-60
- PFA:
-
paraformaldehyde
- BrdU:
-
bromodeoxyuridine
- mPSCs:
-
miniature postsynaptic currents
- NMDA:
-
N-methyl-D-aspartic acid
- AMPA:
-
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- TUNEL:
-
terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end labeling
- OGD:
-
oxygen glucose nutrient deprivation
References
Thom T, Haase N, Rosamond W, Howard VJ, Rumsfeld J, Manolio T et al. Heart disease and stroke statistics – 2006 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2006; 113: e85–e151.
Hicks AU, Lappalainen RS, Narkilahti S, Suuronen R, Corbett D, Sivenius J et al. Transplantation of human embryonic stem cell-derived neural precursor cells and enriched environment after cortical stroke in rats: cell survival and functional recovery. Eur J Neurosci 2009; 29: 562–574.
Alper J . Geron gets green light for human trial of ES cell-derived product. Nat Biotechnol 2009; 27: 213–214.
Wei L, Cui L, Snider BJ, Rivkin M, Yu SS, Lee CS et al. Transplantation of embryonic stem cells overexpressing Bcl-2 promotes functional recovery after transient cerebral ischemia. Neurobiol Dis 2005; 19: 183–193.
Biella G, Di Febo F, Goffredo D, Moiana A, Taglietti V, Conti L et al. Differentiating embryonic stem-derived neural stem cells show a maturation-dependent pattern of voltage-gated sodium current expression and graded action potentials. Neuroscience 2007; 149: 38–52.
Muotri AR, Nakashima K, Toni N, Sandler VM, Gage FH . Development of functional human embryonic stem cell-derived neurons in mouse brain. Proc Natl Acad Sci USA 2005; 102: 18644–18648.
Johnson MA, Weick JP, Pearce RA, Zhang SC . Functional neural development from human embryonic stem cells: accelerated synaptic activity via astrocyte coculture. J Neurosci 2007; 27: 3069–3077.
White BC, Sullivan JM, DeGracia DJ, O’Neil BJ, Neumar RW, Grossman LI et al. Brain ischemia and reperfusion: molecular mechanisms of neuronal injury. J Neurol Sci 2000; 179: 1–33.
Zawada WM, Zastrow DJ, Clarkson ED, Adams FS, Bell KP, Freed CR . Growth factors improve immediate survival of embryonic dopamine neurons after transplantation into rats. Brain Res 1998; 786: 96–103.
McDonald JW, Liu XZ, Qu Y, Liu S, Mickey SK, Turetsky D et al. Transplanted embryonic stem cells survive, differentiate and promote recovery in injured rat spinal cord. Nat Med 1999; 5: 1410–1412.
Daadi MM, Maag AL, Steinberg GK . Adherent self-renewable human embryonic stem cell-derived neural stem cell line: functional engraftment in experimental stroke model. PLoS ONE 2008; 3: e1644.
Darsalia V, Kallur T, Kokaia Z . Survival, migration and neuronal differentiation of human fetal striatal and cortical neural stem cells grafted in stroke-damaged rat striatum. Eur J Neurosci 2007; 26: 605–614.
Modo M, Stroemer RP, Tang E, Patel S, Hodges H . Effects of implantation site of dead stem cells in rats with stroke damage. Neuroreport 2003; 14: 39–42.
Kirino T . Ischemic tolerance. J Cereb Blood Flow Metab 2002; 22: 1283–1296.
Pong K . Ischaemic preconditioning: therapeutic implications for stroke? Expert Opin Ther Targets 2004; 8: 125–139.
Hu X, Yu SP, Fraser JL, Lu Z, Ogle ME, Wang JA et al. Transplantation of hypoxia-preconditioned mesenchymal stem cells improves infarcted heart function via enhanced survival of implanted cells and angiogenesis. J Thorac Cardiovasc Surg 2008; 135: 799–808.
Theus MH, Wei L, Cui L, Francis K, Hu X, Keogh C et al. In vitro hypoxic preconditioning of embryonic stem cells as a strategy of promoting cell survival and functional benefits after transplantation into the ischemic rat brain. Exp Neurol 2008; 210: 656–670.
Ezashi T, Das P, Roberts RM . Low O2 tensions and the prevention of differentiation of hES cells. Proc Natl Acad Sci USA 2005; 102: 4783–4788.
Erecinska M, Silver IA . Tissue oxygen tension and brain sensitivity to hypoxia. Respir Physiol 2001; 128: 263–276.
Xu RH, Chen X, Li DS, Li R, Addicks GC, Glennon C et al. BMP4 initiates human embryonic stem cell differentiation to trophoblast. Nat Biotechnol 2002; 20: 1261–1264.
Ying QL, Nichols J, Chambers I, Smith A . BMP induction of Id proteins suppresses differentiation and sustains embryonic stem cell self-renewal in collaboration with STAT3. Cell 2003; 115: 281–292.
Itsykson P, Ilouz N, Turetsky T, Goldstein RS, Pera MF, Fishbein I et al. Derivation of neural precursors from human embryonic stem cells in the presence of noggin. Mol Cell Neurosci 2005; 30: 24–36.
Draper JS, Smith K, Gokhale P, Moore HD, Maltby E, Johnson J et al. Recurrent gain of chromosomes 17q and 12 in cultured human embryonic stem cells. Nat Biotechnol 2004; 22: 53–54.
Horie N, So K, Moriya T, Kitagawa N, Tsutsumi K, Nagata I et al. Effects of oxygen concentration on the proliferation and differentiation of mouse neural stem cells in vitro. Cell Mol Neurobiol 2008; 28: 833–845.
Studer L, Csete M, Lee SH, Kabbani N, Walikonis J, Wold B et al. Enhanced proliferation, survival, and dopaminergic differentiation of CNS precursors in lowered oxygen. J Neurosci 2000; 20: 7377–7383.
McConnell SK . Migration and differentiation of cerebral cortical neurons after transplantation into the brains of ferrets. Science 1985; 229: 1268–1271.
Arsham AM, Plas DR, Thompson CB, Simon MC . Phosphatidylinositol 3-kinase/Akt signaling is neither required for hypoxic stabilization of HIF-1 alpha nor sufficient for HIF-1-dependent target gene transcription. J Biol Chem 2002; 277: 15162–15170.
Pore N, Jiang Z, Shu HK, Bernhard E, Kao GD, Maity A . Akt1 activation can augment hypoxia-inducible factor-1alpha expression by increasing protein translation through a mammalian target of rapamycin-independent pathway. Mol Cancer Res 2006; 4: 471–479.
Pasha Z, Wang Y, Sheikh R, Zhang D, Zhao T, Ashraf M . Preconditioning enhances cell survival and differentiation of stem cells during transplantation in infarcted myocardium. Cardiovasc Res 2008; 77: 134–142.
Covello KL, Kehler J, Yu H, Gordan JD, Arsham AM, Hu CJ et al. HIF-2alpha regulates Oct-4: effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev 2006; 20: 557–570.
Milosevic J, Maisel M, Wegner F, Leuchtenberger J, Wenger RH, Gerlach M et al. Lack of hypoxia-inducible factor-1 alpha impairs midbrain neural precursor cells involving vascular endothelial growth factor signaling. J Neurosci 2007; 27: 412–421.
Paulding WR, Czyzyk-Krzeska MF . Regulation of tyrosine hydroxylase mRNA stability by protein-binding, pyrimidine-rich sequence in the 3′-untranslated region. J Biol Chem 1999; 274: 2532–2538.
Keogh CL, Yu SP, Wei L . The effect of recombinant human erythropoietin on neurovasculature repair after focal ischemic stroke in neonatal rats. J Pharmacol Exp Ther 2007; 322: 521–528.
Trouillas M, Saucourt C, Duval D, Gauthereau X, Thibault C, Dembele D et al. Bcl2, a transcriptional target of p38alpha, is critical for neuronal commitment of mouse embryonic stem cells. Cell Death Differ 2008; 15: 1450–1459.
Fukuda R, Zhang H, Kim JW, Shimoda L, Dang CV, Semenza GL . HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 2007; 129: 111–122.
Wick A, Wick W, Waltenberger J, Weller M, Dichgans J, Schulz JB . Neuroprotection by hypoxic preconditioning requires sequential activation of vascular endothelial growth factor receptor and Akt. J Neurosci 2002; 22: 6401–6407.
Chavez JC, Baranova O, Lin J, Pichiule P . The transcriptional activator hypoxia inducible factor 2 (HIF-2/EPAS-1) regulates the oxygen-dependent expression of erythropoietin in cortical astrocytes. J Neurosci 2006; 26: 9471–9481.
Baranova O, Miranda LF, Pichiule P, Dragatsis I, Johnson RS, Chavez JC . Neuron-specific inactivation of the hypoxia inducible factor 1 alpha increases brain injury in a mouse model of transient focal cerebral ischemia. J Neurosci 2007; 27: 6320–6332.
Jiang M, Wang B, Wang C, He B, Fan H, Guo TB et al. Angiogenesis by transplantation of HIF-1 alpha modified EPCs into ischemic limbs. J Cell Biochem 2008; 103: 321–334.
Wei L, Ying DJ, Cui L, Langsdorf J, Yu SP . Necrosis, apoptosis and hybrid death in the cortex and thalamus after barrel cortex ischemia in rats. Brain Res 2004; 1022: 54–61.
Acknowledgements
This study was supported by NIH Grants 058710, NS37372 and NS 045810 (L Wei), NIH 1T32ES012878-02 (K Francis).
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by G Melino
Rights and permissions
This article is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 3.0 license. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Francis, K., Wei, L. Human embryonic stem cell neural differentiation and enhanced cell survival promoted by hypoxic preconditioning. Cell Death Dis 1, e22 (2010). https://doi.org/10.1038/cddis.2009.22
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2009.22
Keywords
This article is cited by
-
Human placenta-derived amniotic epithelial cells as a new therapeutic hope for COVID-19-associated acute respiratory distress syndrome (ARDS) and systemic inflammation
Stem Cell Research & Therapy (2022)
-
Core–shell hydrogel microcapsules enable formation of human pluripotent stem cell spheroids and their cultivation in a stirred bioreactor
Scientific Reports (2021)
-
Biomimetic microenvironmental preconditioning enhance neuroprotective properties of human mesenchymal stem cells derived from Wharton's Jelly (WJ-MSCs)
Scientific Reports (2020)
-
Metabolic regulation of neurodifferentiation in the adult brain
Cellular and Molecular Life Sciences (2020)
-
LincRNA-p21 promotes mesenchymal stem cell migration capacity and survival through hypoxic preconditioning
Stem Cell Research & Therapy (2018)
