
Abstract
Intrinsic apoptosis is controlled by the BCL-2 family of proteins but the complexity of intra-family interactions makes it challenging to predict cell fate via standard molecular biology techniques. We discuss BCL-2 family regulation and how to determine cells’ readiness for apoptosis and anti-apoptotic dependence. Cancer cells often adopt anti-apoptotic defense mechanisms in response to oncogenic stress or anti-cancer therapy. However, by determining their anti-apoptotic addiction, we can use novel BH3 mimetics to overwhelm this apoptotic blockade. We outline the development and uses of these unique anti-apoptotic inhibitors and how to possibly combine them with other anti-cancer agents using dynamic BH3 profiling (DBP) to improve personalized cancer treatment.
Similar content being viewed by others

Main

Facts
-
BCL-2 family proteins and their complex interactions regulate intrinsic apoptosis.
-
BH3 profiling can determine cancer cells’ priming for cell death and anti-apoptotic dependencies.
-
Cancer cells often acquire anti-apoptotic defense mechanisms against oncogenic stress and therapy.
-
The DBP functional assay can be used to determine effective combinations of anti-cancer agents with BH3 mimetics to improve cancer treatment.
Open Questions
-
Is anti-apoptotic adaptation a common defense mechanism in most cancer types?
-
Can BH3 mimetics alone or in combination be used to treat solid tumors?
-
Will functional assays be able to guide BH3 mimetics’ use in the clinic?
Programmed cell death (PCD) has been observed in many forms of life from metazoans to mammals. Perhaps the best studied PCD pathway, and the first characterized, is apoptosis. Kerr and collaborators first described apoptosis in the early 70s in mammalian tissue sections where they observed that dying cells showed stereotypic nuclear condensation and cellular fragmentation. Moreover, they found that these fragments were phagocytosed by nearby cells.1 These fragments, also known as apoptotic bodies, are the remains of the plasma membrane, containing cell fragments and presenting phosphatidylserine in their surface as an 'eat me signal' that can be recognized by phagocytic white blood cells.2
Two distinct pathways of apoptosis have been previously characterized: extrinsic and intrinsic. The extrinsic pathway occurs when specific receptors on the cell surface called death receptors are activated, such as TNFR, FAS (CD95) and DR3/WSL. Ligand binding to the receptor induces a change in the intracellular region that promotes adapter proteins activation and death-inducing signaling complex formation. Thus, initiator caspases, such as caspase-8, get cleaved and activated, resulting in initiation of downstream executioner caspases that orchestrate apoptosis. Active caspase-8 also can cleave and activate the BH3-only protein BID that can promote the intrinsic pathway, connecting both apoptotic modes.3
Intrinsic apoptosis, also referred as the mitochondrial pathway, is executed in response to cellular damage and most anti-cancer agents, and the B-cell lymphoma 2 or BCL-2 family proteins regulate it. These proteins control mitochondrial outer membrane permeabilization (MOMP), which for most instances can be considered the point of no return for apoptosis. This permeabilization allows the release of soluble proteins such as cytochrome c and SMAC/DIABLO, from the mitochondrial intermembrane space into the cytosol. Once these proteins are released, cytochrome c binds to APAF-1 and caspase-9 in presence of dATP to form the apoptosome, which activates downstream effector caspases and triggers apoptosis.4
BCL2: the family founder
The founding member of the family, BCL-2 was identified as an oncogene resulting from a translocation between chromosomes 14 and 18 that promoted malignant lymphomagenesis.5, 6 In the early 90’s, several laboratories identified BCL-2 as a pro-survival protein that prevented apoptotic cell death and facilitated MYC-induced transformation.7, 8 The next obvious question at the time was: if BCL-2 protects from PCD, which proteins promote apoptosis? The answer came with the identification of a pro-death protein bound to BCL-2, named BCL-2-associated X or BAX, with similar structure and sequence homology to BCL-2.9 The other members of the so-called BCL-2 family were discovered in the following years based on protein interactions and their similar protein structure. They were classified as pro- or anti-apoptotic depending on their biological activity.10, 11, 12, 13 Following their identification, it became clear that MOMP and the release of cytochrome c was the triggering event of apoptosis and that BCL-2 prevented cell death by stopping this event.14, 15, 16, 17
The BCL2 family of pro-apoptotic and anti-apoptotic proteins
The BCL-2 family proteins can be classified based on their structure and BCL-2 homology (BH) domains. The anti-apoptotic members BCL-2, BCL-XL, BCL-W, MCL-1 and A1/BFL-1 possess four BH domains, BH1-BH4, and present a hydrophobic groove in their structure that binds to the BH3 domain found in the pro-apoptotic. The pro-apoptotic effector proteins, BAX, BAK and BOK, possess three to four BH domains, and have the capacity to form pores in the mitochondrial outer membrane.18, 19, 20 These domains are composed of nine α-helices, including a transmembrane C-terminal α-helix that can anchor to the mitochondrial outer membrane. Their tridimensional structure are very similar to membrane pore-forming domains found in diphtheria toxin and colicins.21 There is a third group of proteins possessing only the BH3 domain, therefore named BH3-only proteins, with a pro-apoptotic function, including BIM, BID, PUMA, BAD, NOXA, HRK, BMF, BIK and others.13, 22, 23, 24, 25, 26, 27 Interaction between pro- and anti-apoptotic proteins takes place via the binding of the hydrophobic face of the amphipathic BH3 alpha-helix from the pro-apoptotic protein into a hydrophobic pocket in the anti-apoptotic protein formed by the BH1, BH2 and BH3 domains.
Regulation of MOMP by the BCL-2 family of proteins
Once they are activated, effector proteins BAX and BAK oligomerize in the mitochondrial outer membrane to promote MOMP and cytochrome c release.18, 19 In fact, a double knockout of BAX and BAK prevents apoptosis in response to most stimuli.28, 29 The third effector protein BOK may be regulated by protein stability and can induce MOMP in response to endoplasmic reticulum stress insults, and its activation may not rely on other BCL-2 family proteins.30 However, regulation of BOK remains a matter of uncertainty.31, 32 BAX and BAK oligomerization can be directly engaged by activator BH3-only proteins such as BIM13, 33 and BID,22 although showing specificity differences.34 Other BH3-only proteins have recently emerged as direct activators as well, such as PUMA and NOXA, although with less potency compared to BIM or BID.35, 36, 37, 38 The BH3 domains found in these BH3-only proteins induce an allosteric change in BAX and BAK that promotes the formation of high-ordered oligomers that form pores in the mitochondrial outer membrane to cause MOMP.39, 40, 41
The anti-apoptotic proteins, including BCL-2, BCL-XL, MCL-1, BCL-W and BFL-1/A1, can prevent MOMP and apoptosis by two complementary mechanisms. They can directly bind the BH3 domain of activator BH3-only proteins, preventing BAX and BAK oligomerization.42, 43, 44 On the other hand, they can directly bind the activated monomeric forms of BAX and BAK, whose conformational change induces BH3 domain exposure.45, 46, 47 The most potent activators’ BH3 domains, like BIM, BID (and PUMA), interact promiscuously with all anti-apoptotic proteins and can directly activate BAX and BAK.
The sensitizers within the family and their mechanism of action
There is a fourth group of BCL-2 family proteins possessing only a BH3 domain, the so-called sensitizers. Sensitizers can be considered to include PUMA, BAD, HRK, NOXA, BMF and BIK. A pure sensitizer would not be able to directly activate BAX and BAK but rather exert pro-apoptotic effect by competing for binding in the hydrophobic pocket of the anti-apoptotic BCL-2 family members.42, 44, 48 It has become clear that there is something of a continuum between pure activator and pure sensitizer function, with BIM and BID being the most potent activators at one end, and others like PUMA possessing detectable but likely less-potent activity. In brief, the sensitizer BH3 domain binds to anti-apoptotic proteins, displacing activators that can activate the effectors and induce MOMP, exerting an indirect pro-apoptotic function.42 Alternatively, sensitizers can displace monomeric activated BAX or BAK that are sequestered by an anti-apoptotic protein. Sensitizers’ BH3 sequence governs their affinity for anti-apoptotic proteins. For example, BAD has high affinity for BCL-2, BCL-XL and BCL-W, but not MCL-1 or BFL-1. In contrast, HRK selectively binds to BCL-XL, and NOXA specifically binds to MCL-1 42 (Figure 1). Therefore, the biological effect of sensitizers greatly depends upon the pre-existing balance of other BCL-2 family members at the mitochondria.44, 49

The BCL-2 family of proteins. BCL-2 family control over mitochondrial apoptosis representation. In response to therapy or oncogene activation, activators engage effectors, causing mitochondrial permeabilization and apoptotic cell death. PUMA and NOXA, are weaker activators compared with BIM or BID. Anti-apoptotic proteins sequester activators or effector proteins to prevent apoptosis. Sensitizers act as selective antagonists of anti-apoptotic proteins. Modified from Deng et al., 2007
Apoptosis, BH3 Mimetics and Cancer
Are cancer cells more prone to apoptosis?
Why does chemotherapy work? Despite the broad use of anti-cancer agents in cancer treatment, and their ability to cure certain cancers, why chemotherapy selectively kills cancer cells and spares normal cells is poorly understood. Only recently Sarosiek and collaborators described how adult tissues are refractory for apoptosis, with low expression of apoptotic proteins in vital organs like brain, heart, kidney and liver, but not in hematopoietic tissues or intestine.50, 51 Chemo-sensitive cancer cells are more disposed for apoptosis than chemo-resistant cancer cells or normal cells, observations that likely elucidate why there is a therapeutic window to clinically treat tumors with chemotherapy.50
Cancer cells die by apoptosis when exposed to the right therapy. This apoptotic cell death can occur by inducing an up regulation of pro-apoptotic proteins or by directly decreasing the anti-apoptotic reservoir to allow the activators to initiate MOMP. Most anti-cancer agents like targeted therapies, DNA-damaging agents, microtubule inhibitors or other cytotoxic molecules, produce a cellular stress that leads to cell death.52, 53 This stress often primes by increasing expression of activators that can engage BAX/BAK to induce MOMP and apoptosis unless sequestered by anti-apoptotic proteins.34, 52, 54, 55, 56, 57, 58 Despite many successes in cancer therapy, many tumors are not efficiently killed by chemotherapy, leading some to suggest that cancer cells are generally blocked in apoptotic signaling.51, 59 It is important to ask, 'Compared to what?'. Many laboratories have observed that cells undergoing oncogenic transformation exhibit higher expression of pro-apoptotic proteins resulting from cell cycle checkpoint evasion, DNA replication stress, unfolded protein response or oxidative stress. However, cancer cells nonetheless survive by blocking these signals,60, 61 leaving a cell that is rather tenuously dependent for survival on continuation of that block. Rather than leaving a cell that is fundamentally prevented from undergoing apoptosis, very often the resulting cancer cell is quite ready to undergo apoptosis compared to normal cells. This distinction is, indeed, an important determinant of the therapeutic index for conventional chemotherapy that we have benefitted from for decades.50, 62
Mechanisms of anti-apoptotic adaptation
One way that cancer cells can escape death from the pro-apoptotic signaling that emerges in oncogenesis or therapy is via selection for higher expression of anti-apoptotic proteins. Therefore, even if the cytotoxic insult is effective to induce a pro-death signaling by upregulating activator proteins like BIM or directly activating BAX or BAK, cancer cells can survive by sequestering them with anti-apoptotic proteins. This anti-apoptotic adaptation can vary from tumor to tumor, therefore it is necessary to evaluate it for every case.49 For instance, different cases of multiple myeloma (MM) can rely on BCL-2, BCL-XL, MCL-1 or combinations thereof for their survival, pointing to the need of case-specific treatments.63 In contrast, T-cell acute lymphoblastic leukemia anti-apoptotic dependency changes with the differentiation stage of the leukemic clone, relying largely on BCL-XL except for early T-cell progenitor cells that are BCL-2 dependent.64 Moreover, many laboratories have characterized tumor evolution and anti-apoptotic adaptation in response to therapy,53, 65, 66, 67, 68, 69 demanding rapid ways to measure it that will be discussed below. This state in which cancer cells constrain pro-apoptotic proteins via heterodimerization with anti-apoptotic proteins can be termed being ‘primed’ for apoptosis. In these cases, the anti-apoptotic defenses can be overwhelmed by blocking the anti-apoptotic proteins with specific inhibitors.67, 70, 71, 72, 73, 74
Tackling anti-apoptotic adaptation
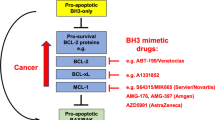
Consequently, great efforts have been directed towards finding small molecules to inhibit these anti-apoptotic BCL-2 family proteins and promote apoptosis in cancer with the so-called BH3 mimetics that mimic the action of certain BH3-only proteins. The first demonstration that abrogation of function of an anti-apoptotic protein by itself could lead to cancer regression was via the use of regulatable expression of BCL-2 in a murine model of leukemia.75 Later work found that deletion of MCL-1 caused regression of a similar MYC-driven lymphoma.58 The first attempt to inhibit the anti-apoptotic proteins was described in 2000 with the discovery of HA14-1, a small molecule targeting the BCL-2 surface pocket that showed activity in vitro.76 A subsequent effort by others produced obatoclax (GX-15-070), a drug that showed relatively low affinity,77 a BAX/BAK-independent induction of cell death 78 and modest clinical activity.79 Abbott Laboratories (Abbott Park, IL, USA) developed a small molecule matching the binding pattern of BAD, inhibiting BCL-2/BCL-XL/BCL-W, named ABT-737.42, 80 Preclinical studies with ABT-737 showed on-target activity and a requirement for BAX and BAK to induce apoptosis.81
Abbott Laboratories (now Abbvie) developed an orally bioavailable analog called ABT-263 (navitoclax) for clinical use. Preclinical studies with both ABT-737 and ABT-263 demonstrated their mechanism of action, displacing pro-apoptotic BH3-only proteins from BCL-2.73 When both agents were tested in vivo, a decrease in platelet counts owing to BCL-XL inhibition was observed, impairing ABT-263 use in the clinic.70, 82, 83, 84 However, ABT-263 related thrombocytopenia can be controlled using appropriate dosing85, 86 and several clinical trials evaluating its safety and efficacy are currently ongoing in CLL, small-cell lung cancer and other solid tumors87, 88 (see https://clinicaltrials.gov/ct2/results?term=abt-263). Encouraged by some striking responses in CLL patients to this BH3 mimetic, Abbvie developed an orally available inhibitor with 100-fold greater affinity for BCL-2 than BCL-XL, named ABT-199 (venetoclax).89 Owing to its high subnanomolar affinity for BCL-2 and low binding to BCL-XL, this novel agent does not cause thrombocytopenia. In fact, venetoclax was such a potent BCL-2 inhibitor that it caused tumor lysis syndrome in some CLL patients, imposing a dose-escalation strategy and the need for close surveillance in the clinic.90 The single-agent activity of venetoclax in relapsed CLL patients was outstanding with an 80% response rate and manageable secondary effects.90 Clinical trial results of venetoclax as a single agent led to its FDA approval in April 2016 for its use in CLL patients with 17p chromosomal deletion, a biomarker for TP53 loss and poor prognosis.71, 90, 91 Consequently, venetoclax became the first BH3 mimetic approved by the FDA for cancer treatment, encouraging many to follow.
Since then, a myriad of clinical trials started to evaluate this agent alone or in combination to treat multiple hematological malignancies (https://clinicaltrials.gov/ct2/results?term=abt-199). In contrast to relatively homogeneous results in CLL, other blood cancers like acute myeloid leukemia (AML), MM and non-Hodgkin’s lymphoma showed heterogeneous responses to single-agent venetoclax.89, 92 To improve response to this agent, many studies assessed its combination with other chemotherapeutic drugs. For instance, in AML the combination of venetoclax with hypomethylating agents results in an improvement of its clinical activity, achieving ~70% response rate (combined CR and CRi), in patients.93 In solid tumors, several reports showed in vitro response to venetoclax in breast67 and small-cell lung carcinoma lines,80 but only in certain cell lines, pointing to a need for stratification.
Following ABT-199 success, several pharmaceutical companies, including Servier, AstraZeneca and Amgen, are developing novel BCL-2, BCL-XL and MCL-1 inhibitors, and evaluating them in liquid and solid tumors,94 (https://clinicaltrials.gov/ct2/results?term=mcl-1+OR+bcl-2+OR+bcl-XL+inhibitor). Selective BCL-XL inhibition could be useful to treat certain types of tumors, as they upregulate it as a mechanism of protection against apoptosis.95, 96 However, BCL-XL dependence is found only in certain cases, pointing to an unmet need for predictive biomarkers for patient selection.97
There is evidence for the efficacy of MCL-1 inhibitors. However, although CLL is an example of a disease quite homogeneously dependent on BCL-2, a cancer homogeneously dependent on MCL-1 has yet to be identified. Heterogeneous MCL-1 dependence has been observed in non-small cell lung cancer lines, AML, chronic myelogenous leukemia, B-cell acute lymphoblastic leukemia (B-ALL) and MM.66, 98, 99, 100, 101 MCL-1 inhibition may have side effects like hematopoietic toxicity102 cardiotoxicity103 and perhaps affect mitochondrial respiration.104 However, it remains to be seen the extent to which titratable MCL-1 inhibition by drugs in humans phenocopies gene deletion in mice. As functions for MCL-1 not depending on BH3 domain binding have been identified, it is possible that some effects of MCL-1 deletion observed in mouse models may be separable from those caused by BH3 mimetic inhibition.104 Several MCL-1 inhibitors have been developed over the years using different strategies showing modest selectivity.105, 106, 107 However, the novel S63845 molecule developed by Servier exhibits exquisite selectivity for MCL-1 over BCL-2 and BCL-XL, and shows promising results.72, 108
Furthermore, based on the apoptotic resistance observed in vital organs in adults,51 there should theoretically be a therapeutic index to block all anti-apoptotic proteins by combining several BH3 mimetics. In fact, pre-clinical experiments showed that BCL-2/XL inhibition may lead to MCL-1 increased expression65 or BCL-XL109, 110 as a compensatory survival adaptation. Therefore, using a pan-BCL-2 inhibitor strategy to treat the most aggressive types of cancer may have clinical benefit. Like other chemotherapies used in the clinic, this therapeutic strategy would affect organs that are more ‘primed’ for apoptosis like the hematopoietic system, leading to well-characterized secondary effects. However, the clinical experience with myelosuppression lends optimism that hematologic toxicity can be managed via dosing, timing and combination of agents, and by closely monitoring patients. Moreover, should bone marrow toxicity be unavoidable, efficacious therapies can be made tolerable using autologous and allogeneic stem cell transplantation techniques. The clinical utility of these new inhibitors suggests that we will see an expansion of their use in the next years.
Directing BH3 Mimetics in the Clinic
The dawn of BH3 mimetics brings an important question: how can we determine when to better use them to treat cancer patients? As discussed above, the BCL-2 family of proteins includes over a dozen members, each presenting different mode of regulation, post-translational modification, and protein–protein interactions, making difficult a correct prediction for BH3 mimetics’ response in cancer cells using standard molecular biology techniques. In other words, we have excellent agents to block anti-apoptotic defense in cancer cells but we lack efficient predictive biomarkers to guide their use. So far, most clinical successes with BH3 mimetics have arisen from exploiting lineage-based, rather than genome-based, vulnerabilities, so that genetic biomarkers have not been useful in predicting response. Therefore, there is a clear unmet need for novel ways to predict clinical efficacy of BH3 mimetics.
Identifying cancer anti-apoptotic addiction through BH3 profiling
In this regard, the Letai laboratory developed a technique called BH3 profiling that can determine tumors’ dependencies on individual anti-apoptotic proteins.42, 48, 111, 112, 113 BH3 profiling expose cancer cells’ mitochondria to synthetic 20-mer BH3 peptides that mimic the pro-apoptotic function of BH3-only proteins to determine MOMP and apoptosis initiation.44, 112, 113 For instance, BIM and BID BH3 peptides interact with all endogenous anti-apoptotic proteins and can activate the effectors BAX and BAK, and are used as a measure of overall apoptotic priming.34, 42, 51 In contrast, the synthetic PUMA BH3 peptide binds with all the anti-apoptotic proteins but cannot activate BAX or BAK.42 The information obtained by BH3 profiling using these peptides is useful for identifying how close a cancer cell is to the apoptotic threshold, or how ’primed’ for apoptosis, and has proved to be a good predictor of response to conventional chemotherapy.50, 51, 62 Interestingly, certain BH3 peptides interact selectively with endogenous anti-apoptotic BCL-2 family proteins and can guide the therapeutic use of BH3 mimetics.42, 48 For example, MS1 and NOXA BH3 peptides interact selectively with MCL-1,114 the HRK BH3 peptide interacts selectively with BCL-XL, the BAD BH3 peptide interacts selectively with both BCL-2 and BCL-XL.42 By using these anti-apoptotic selective peptides, the Letai lab found BCL-2 dependence in CLL;74 heterogenous anti-apoptotic dependence in AML 62, 115 and MM;63 BCL-2 dependence in B-cell acute lymphoblastic leukemia and blastic plasmacytoid dendritic cell neoplasm;73, 116, 117 BCL-XL dependence in T-cell acute lymphoblastic leukemia, but BCL-2 dependence in early T-cell progenitor cell leukemia.64, 118
Dynamic BH3 profiling (DBP)
Most anti-cancer agents, including kinase inhibitors, induce apoptosis in cells that are dependent on a pathway targeted by the inhibitor. When a cancer cell is treated with a targeted agent, early changes in the BCL-2 family of proteins can be rapidly detected preceding the activation of apoptosis. Taking advantage of this biological observation, the Letai laboratory developed a novel functional assay called DBP that measures net changes in mitochondrial apoptotic signaling in cancer cells following administration of targeted therapeutic agents using titrated doses of BIM BH3 peptide. DBP can reveal drug treatments that increase priming in cancer cells (termed Δ% priming), which is predictive of their later cell death in vitro, in vivo and directly in patient samples119 (Figure 2). This technology has been successfully applied to find new treatments for AML,120 B-ALL,121, 122 and to determine venetoclax response in blastic plasmacytoid dendritic cell neoplasm.117

The threshold of apoptosis. We define priming as the proximity of a malignant cell to the threshold of apoptosis, and dynamic BH3 profiling (DBP) measures increases in priming upon treatment. (a) When a cancer cell is exposed to the wrong treatment there is no increase in priming, giving a negative result for DBP, and the cell will survive. (b) In contrast, when a cancer cell is exposed to the right treatment there is an increase in priming that will be detected by DBP in early timepoints preceding apoptotic cell death
Combining BH3 Mimetics with Other Treatments
Although we discussed clear examples in where BH3 mimetics can be useful as single agents to treat different types of cancer, many scientific reports suggest their therapeutic use in combination with other anti-cancer agents. For example, combining navitoclax with gemcitabine to treat solid tumors,123 navitoclax with brentuximab to treat Hodgkin’s lymphoma,124 JAK2 inhibitors with ABT-737125 or ABT-737 with cisplatin to treat non-small cell lung cancer.126 Particularly interesting are the possible combinations of targeted therapies with BH3 mimetics.68, 126, 127, 128, 129 In fact, dozens of clinical trials are currently evaluating the combined use of BH3 mimetics with other chemotherapeutic agents to treat multiple hematological malignancies (see clinicaltrials.gov). As discussed above, cancer cells’ anti-apoptotic dependence can alter when exposed to therapy by increasing the availability or upregulating anti-apoptotic proteins.54, 56, 59 By studying how this adaptation occurs, we can design novel therapeutic strategies combining anti-cancer agents and BH3 mimetics to restore cancer’s cell death (Figure 3).

Use of BH3 mimetics to overcome tumors’ resistance to therapy. When cancer cells are exposed to therapy, they can adapt their anti-apoptotic strategy to block the pro-apoptotic proteins’ accumulation (activators and active effectors) induced by treatment. By blocking the anti-apoptotic defense using a specific BH3 mimetic, activators will be displaced from anti-apoptotic proteins and restore apoptotic cell death
This brings us to our initial question: how can we determine when to use BH3 mimetics, and in what combinations, to treat cancer patients? In this regard, we can perform DBP, challenging cancer cells with active agents of interest, and then use specific peptides like BAD, HRK or NOXA to determine increased dependence on BCL-2, BCL-XL or MCL-1. We can analyze this information to design rational therapeutic strategies exploiting cancer’s anti-apoptotic vulnerabilities. For example, we can examine combinations of agents like kinase inhibitors or conventional chemotherapy with BH3 mimetics to treat cancer patients. These drugs can be used together, or be given sequentially in a metronomic regime to increase potency and minimize secondary effects, and DBP can evaluate many possibilities to guide precision medicine (Figure 4). In fact, this strategy has already been successfully applied in chronic lymphocytic leukemia patient samples by combining the BTK inhibitor ibrutinib, which led to increased BCL-2 dependence, with venetoclax to enhance cancer cells’ killing.127 We and others believe that the next years will witness an expansion of similar strategies to treat different types of cancer, both liquid and solid.

Using dynamic BH3 profiling to identify BH3 mimetics’ use. To perform dynamic BH3 profiling, we obtain a single cell suspension from a cell line or a primary sample, and we expose the cells for a short incubation with the different treatments of interest. After this incubation, we permeabilize cells with digitonin, and expose the cells to selective BH3 peptides. We then analyze cytochrome c retention in the cells by flow cytometry or microscopy. By comparing non-treated with treated cells, we will determine the Δ%priming for each agent and identify which are most effective to induce apoptosis or detect anti-apoptotic dependency changes. BIM BH3 peptide will indicate overall response to treatment (treatment 1). A BAD BH3-positive signal and negative HRK signal will indicate BCL-2 dependence and a possible use for agents like ABT-199/venetoclax (treatment 2). HRK-positive signal will indicate BCL-XL dependence and possible use for inhibitors against this antiapoptotic protein (treatment 3). NOXA-positive signal will indicate the use of specific MCL-1 inhibitors (treatment 4). Adapted from Montero et al., 2015 and Ryan et al., 2016
Future Perspectives
As discussed above, many reports showed that cancer cells often present anti-apoptotic adaptation to ensure survival against oncogenic stress or anti-cancer therapy. Even if this adaptation has been observed in many malignancies, it is still unknown if this is a common feature for all human cancers or if on the contrary it is observed more heterogeneously. With FDA-approval of venetoclax in CLL, many oncologists and cancer researchers are exploring the uses of BH3 mimetics in other contexts in the clinic. Particularly, in hematological malignancies a myriad of clinical trials are assessing the possible use of venetoclax alone or in combination with other chemotherapies. Furthermore, several pharmaceutical companies are developing other BH3 mimetics targeting not only BCL-2, but also BCL-XL and MCL-1. It remains to be seen whether the excellent results observed in diseases like CLL using anti-apoptotic inhibition will be mirrored in solid tumors that are commonly more difficult to treat in the clinic. The authors expect that a diversity of BH3 mimetics will need to be employed, with likely a greater importance for BCL-XL inhibition in solid tumors than has so far been observed in hematologic tumors. One of the biggest problems for the use of BH3 mimetics is the lack of specific biomarkers to guide their use. We believe that functional assays like BH3 profiling or DBP may be useful to determine their best use. Undoubtedly, the use of BH3 mimetics in the clinic represents the dawn of a new era for personalized cancer treatment with great promise to improve patients’ clinical outcome.
References
Kerr JF, Wyllie AH, Currie AR . Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 1972; 26: 239–257.
Elmore S . Apoptosis: a review of programmed cell death. Toxicol Pathol 2007; 35: 495–516.
Green DR . Cell death and the immune system: getting to how and why. Immunol Rev 2017; 277: 4–8.
Zou H, Li Y, Liu X, Wang X . An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J Biol Chem 1999; 274: 11549–11556.
McDonnell TJ, Korsmeyer SJ . Progression from lymphoid hyperplasia to high-grade malignant lymphoma in mice transgenic for the t(14; 18). Nature 1991; 349: 254–256.
Vaux DL, Cory S, Adams JM . Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature 1988; 335: 440–442.
Hockenbery D, Nunez G, Milliman C, Schreiber RD, Korsmeyer SJ . Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature 1990; 348: 334–336.
Strasser A, Harris AW, Bath ML, Cory S . Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature 1990; 348: 331–333.
Oltvai ZN, Milliman CL, Korsmeyer SJ . Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell 1993; 74: 609–619.
Boise LH, Gonzalez-Garcia M, Postema CE, Ding L, Lindsten T, Turka LA et al. bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell 1993; 74: 597–608.
Kozopas KM, Yang T, Buchan HL, Zhou P, Craig RW . MCL1, a gene expressed in programmed myeloid cell differentiation, has sequence similarity to BCL2. Proc Natl Acad Sci USA 1993; 90: 3516–3520.
Lin EY, Orlofsky A, Wang HG, Reed JC, Prystowsky MB . A1, a Bcl-2 family member, prolongs cell survival and permits myeloid differentiation. Blood 1996; 87: 983–992.
O'Connor L, Strasser A, O'Reilly LA, Hausmann G, Adams JM, Cory S et al. Bim: a novel member of the Bcl-2 family that promotes apoptosis. EMBO J 1998; 17: 384–395.
Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD . The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science 1997; 275: 1132–1136.
Liu X, Kim CN, Yang J, Jemmerson R, Wang X . Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 1996; 86: 147–157.
Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J et al. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science 1997; 275: 1129–1132.
Bossy-Wetzel E, Newmeyer DD, Green DR . Mitochondrial cytochrome c release in apoptosis occurs upstream of DEVD-specific caspase activation and independently of mitochondrial transmembrane depolarization. EMBO J 1998; 17: 37–49.
Wei MC, Lindsten T, Mootha VK, Weiler S, Gross A, Ashiya M et al. tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev 2000; 14: 2060–2071.
Gross A, Jockel J, Wei MC, Korsmeyer SJ . Enforced dimerization of BAX results in its translocation, mitochondrial dysfunction and apoptosis. EMBO J 1998; 17: 3878–3885.
Carpio MA, Michaud M, Zhou W, Fisher JK, Walensky LD, Katz SG . BCL-2 family member BOK promotes apoptosis in response to endoplasmic reticulum stress. Proc Natl Acad Sci USA 2015; 112: 7201–7206.
Muchmore SW, Sattler M, Liang H, Meadows RP, Harlan JE, Yoon HS et al. X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death. Nature 1996; 381: 335–341.
Wang K, Yin XM, Chao DT, Milliman CL, Korsmeyer SJ . BID: a novel BH3 domain-only death agonist. Genes Dev 1996; 10: 2859–2869.
Nakano K, Vousden KH . PUMA, a novel pro-apoptotic gene, is induced by p53. Mol Cell 2001; 7: 683–694.
Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B . PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell 2001; 7: 673–682.
Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T et al. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 2000; 288: 1053–1058.
Yang E, Zha J, Jockel J, Boise LH, Thompson CB, Korsmeyer SJ . Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell 1995; 80: 285–291.
Inohara N, Ding L, Chen S, Nunez G . harakiri, a novel regulator of cell death, encodes a protein that activates apoptosis and interacts selectively with survival-promoting proteins Bcl-2 and Bcl-X(L). EMBO J 1997; 16: 1686–1694.
Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ et al. Pro-apoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 2001; 292: 727–730.
Lindsten T, Ross AJ, King A, Zong WX, Rathmell JC, Shiels HA et al. The combined functions of pro-apoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol Cell 2000; 6: 1389–1399.
Llambi F, Wang YM, Victor B, Yang M, Schneider DM, Gingras S et al. BOK is a non-canonical BCL-2 family effector of apoptosis regulated by ER-associated degradation. Cell 2016; 165: 421–433.
Fernandez-Marrero Y, Ke F, Echeverry N, Bouillet P, Bachmann D, Strasser A et al. Is BOK required for apoptosis induced by endoplasmic reticulum stress? Proc Natl Acad Sci USA 2016; 113: E492–E493.
Ke F, Voss A, Kerr JB, O'Reilly LA, Tai L, Echeverry N et al. BCL-2 family member BOK is widely expressed but its loss has only minimal impact in mice. Cell Death Differ 2012; 19: 915–925.
Merino D, Giam M, Hughes PD, Siggs OM, Heger K, O'Reilly LA et al. The role of BH3-only protein Bim extends beyond inhibiting Bcl-2-like prosurvival proteins. J Cell Biol 2009; 186: 355–362.
Sarosiek KA, Chi X, Bachman JA, Sims JJ, Montero J, Patel L et al. BID preferentially activates BAK while BIM preferentially activates BAX, affecting chemotherapy response. Mol Cell 2013; 51: 751–765.
Chen HC, Kanai M, Inoue-Yamauchi A, Tu HC, Huang Y, Ren D et al. An interconnected hierarchical model of cell death regulation by the BCL-2 family. Nat Cell Biol 2015; 17: 1270–1281.
Dai H, Smith A, Meng XW, Schneider PA, Pang YP, Kaufmann SH . Transient binding of an activator BH3 domain to the Bak BH3-binding groove initiates Bak oligomerization. J Cell Biol 2011; 194: 39–48.
Kim H, Rafiuddin-Shah M, Tu HC, Jeffers JR, Zambetti GP, Hsieh JJ et al. Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat Cell Biol 2006; 8: 1348–1358.
Du H, Wolf J, Schafer B, Moldoveanu T, Chipuk JE, Kuwana T . BH3 domains other than Bim and Bid can directly activate Bax/Bak. J Biol Chem 2011; 286: 491–501.
Antonsson B, Montessuit S, Sanchez B, Martinou JC . Bax is present as a high molecular weight oligomer/complex in the mitochondrial membrane of apoptotic cells. J Biol Chem 2001; 276: 11615–11623.
Czabotar PE, Westphal D, Dewson G, Ma S, Hockings C, Fairlie WD et al. Bax crystal structures reveal how BH3 domains activate Bax and nucleate its oligomerization to induce apoptosis. Cell 2013; 152: 519–531.
Mikhailov V, Mikhailova M, Degenhardt K, Venkatachalam MA, White E, Saikumar P . Association of Bax and Bak homo-oligomers in mitochondria. Bax requirement for Bak reorganization and cytochrome c release. J Biol Chem 2003; 278: 5367–5376.
Certo M, Del Gaizo Moore V, Nishino M, Wei G, Korsmeyer S, Armstrong SA et al. Mitochondria primed by death signals determine cellular addiction to anti-apoptotic BCL-2 family members. Cancer Cell 2006; 9: 351–365.
Cheng EH, Wei MC, Weiler S, Flavell RA, Mak TW, Lindsten T et al. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol Cell 2001; 8: 705–711.
Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ . Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2002; 2: 183–192.
Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI et al. Pro-apoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev 2005; 19: 1294–1305.
Willis SN, Fletcher JI, Kaufmann T, van Delft MF, Chen L, Czabotar PE et al. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science 2007; 315: 856–859.
Ding J, Mooers BH, Zhang Z, Kale J, Falcone D, McNichol J et al. After embedding in membranes anti-apoptotic Bcl-XL protein binds both Bcl-2 homology region 3 and helix 1 of pro-apoptotic Bax protein to inhibit apoptotic mitochondrial permeabilization. J Biol Chem 2014; 289: 11873–11896.
Deng J, Carlson N, Takeyama K, Dal Cin P, Shipp M, Letai A . BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell 2007; 12: 171–185.
Brunelle JK, Letai A . Control of mitochondrial apoptosis by the Bcl-2 family. J Cell Sci 2009; 122: 437–441.
Ni Chonghaile T, Sarosiek KA, Vo TT, Ryan JA, Tammareddi A, Moore Vdel G et al. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science 2011; 334: 1129–1133.
Sarosiek KA, Fraser C, Muthalagu N, Bhola PD, Chang W, McBrayer SK et al. Developmental regulation of mitochondrial apoptosis by c-myc governs age- and tissue-specific sensitivity to cancer therapeutics. Cancer Cell 2017; 31: 142–156.
Faber AC, Li D, Song Y, Liang MC, Yeap BY, Bronson RT et al. Differential induction of apoptosis in HER2 and EGFR addicted cancers following PI3K inhibition. Proc Natl Acad Sci USA 2009; 106: 19503–19508.
Winter PS, Sarosiek KA, Lin KH, Meggendorfer M, Schnittger S, Letai A et al. RAS signaling promotes resistance to JAK inhibitors by suppressing BAD-mediated apoptosis. Sci Signal 2014; 7: ra122.
Bender A, Opel D, Naumann I, Kappler R, Friedman L, von Schweinitz D et al. PI3K inhibitors prime neuroblastoma cells for chemotherapy by shifting the balance towards pro-apoptotic Bcl-2 proteins and enhanced mitochondrial apoptosis. Oncogene 2011; 30: 494–503.
Ren D, Tu HC, Kim H, Wang GX, Bean GR, Takeuchi O et al. BID, BIM, and PUMA are essential for activation of the BAX- and BAK-dependent cell death program. Science 2010; 330: 1390–1393.
Cragg MS, Kuroda J, Puthalakath H, Huang DC, Strasser A . Gefitinib-induced killing of NSCLC cell lines expressing mutant EGFR requires BIM and can be enhanced by BH3 mimetics. PLoS Med 2007; 4: 1681–1689 discussion 90.
Deng J, Shimamura T, Perera S, Carlson NE, Cai D, Shapiro GI et al. Pro-apoptotic BH3-only BCL-2 family protein BIM connects death signaling from epidermal growth factor receptor inhibition to the mitochondrion. Cancer Res 2007; 67: 11867–11875.
Kelly GL, Grabow S, Glaser SP, Fitzsimmons L, Aubrey BJ, Okamoto T et al. Targeting of MCL-1 kills MYC-driven mouse and human lymphomas even when they bear mutations in p53. Genes Dev 2014; 28: 58–70.
Hata AN, Yeo A, Faber AC, Lifshits E, Chen Z, Cheng KA et al. Failure to induce apoptosis via BCL-2 family proteins underlies lack of efficacy of combined MEK and PI3K inhibitors for KRAS-mutant lung cancers. Cancer Res 2014; 74: 3146–3156.
Hanahan D, Weinberg RA . The hallmarks of cancer. Cell 2000; 100: 57–70.
Hanahan D, Weinberg RA . Hallmarks of cancer: the next generation. Cell 2011; 144: 646–674.
Vo TT, Ryan J, Carrasco R, Neuberg D, Rossi DJ, Stone RM et al. Relative mitochondrial priming of myeloblasts and normal HSCs determines chemotherapeutic success in AML. Cell 2012; 151: 344–355.
Touzeau C, Ryan J, Guerriero J, Moreau P, Chonghaile TN, Le Gouill S et al. BH3 profiling identifies heterogeneous dependency on Bcl-2 family members in multiple myeloma and predicts sensitivity to BH3 mimetics. Leukemia 2016; 30: 761–764.
Chonghaile TN, Roderick JE, Glenfield C, Ryan J, Sallan SE, Silverman LB et al. Maturation stage of T-cell acute lymphoblastic leukemia determines BCL-2 versus BCL-XL dependence and sensitivity to ABT-199. Cancer Discov 2014; 4: 1074–1087.
Yecies D, Carlson NE, Deng J, Letai A . Acquired resistance to ABT-737 in lymphoma cells that up-regulate MCL-1 and BFL-1. Blood 2010; 115: 3304–3313.
Koss B, Morrison J, Perciavalle RM, Singh H, Rehg JE, Williams RT et al. Requirement for anti-apoptotic MCL-1 in the survival of BCR-ABL B-lineage acute lymphoblastic leukemia. Blood 2013; 122: 1587–1598.
Vaillant F, Merino D, Lee L, Breslin K, Pal B, Ritchie ME et al. Targeting BCL-2 with the BH3 mimetic ABT-199 in estrogen receptor-positive breast cancer. Cancer Cell 2013; 24: 120–129.
Potter DS, Galvin M, Brown S, Lallo A, Hodgkinson CL, Blackhall F et al. Inhibition of PI3K/BMX cell survival pathway sensitizes to BH3 mimetics in SCLC. Mol Cancer Ther 2016; 15: 1248–1260.
Aichberger KJ, Mayerhofer M, Krauth MT, Skvara H, Florian S, Sonneck K et al. Identification of mcl-1 as a BCR/ABL-dependent target in chronic myeloid leukemia (CML): evidence for cooperative antileukemic effects of imatinib and mcl-1 antisense oligonucleotides. Blood 2005; 105: 3303–3311.
Vogler M, Hamali HA, Sun XM, Bampton ET, Dinsdale D, Snowden RT et al. BCL2/BCL-X(L) inhibition induces apoptosis, disrupts cellular calcium homeostasis, and prevents platelet activation. Blood 2011; 117: 7145–7154.
Anderson MA, Deng J, Seymour JF, Tam C, Kim SY, Fein J et al. The BCL2 selective inhibitor venetoclax induces rapid onset apoptosis of CLL cells in patients via a TP53-independent mechanism. Blood 2016; 127: 3215–3224.
Kotschy A, Szlavik Z, Murray J, Davidson J, Maragno AL, Le Toumelin-Braizat G et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 2016; 538: 477–482.
Del Gaizo Moore V, Schlis KD, Sallan SE, Armstrong SA, Letai A . BCL-2 dependence and ABT-737 sensitivity in acute lymphoblastic leukemia. Blood 2008; 111: 2300–2309.
Del Gaizo Moore V, Brown JR, Certo M, Love TM, Novina CD, Letai A . Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737. J Clin Invest 2007; 117: 112–121.
Letai A, Sorcinelli MD, Beard C, Korsmeyer SJ . Anti-apoptotic BCL-2 is required for maintenance of a model leukemia. Cancer Cell 2004; 6: 241–249.
Wang JL, Liu D, Zhang ZJ, Shan S, Han X, Srinivasula SM et al. Structure-based discovery of an organic compound that binds Bcl-2 protein and induces apoptosis of tumor cells. Proc Natl Acad Sci USA 2000; 97: 7124–7129.
Nguyen M, Marcellus RC, Roulston A, Watson M, Serfass L, Murthy Madiraju SR et al. Small molecule obatoclax (GX15-070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. Proc Natl Acad Sci USA 2007; 104: 19512–19517.
Konopleva M, Watt J, Contractor R, Tsao T, Harris D, Estrov Z et al. Mechanisms of antileukemic activity of the novel Bcl-2 homology domain-3 mimetic GX15-070 (obatoclax). Cancer Res 2008; 68: 3413–3420.
Paik PK, Rudin CM, Pietanza MC, Brown A, Rizvi NA, Takebe N et al. A phase II study of obatoclax mesylate, a Bcl-2 antagonist, plus topotecan in relapsed small cell lung cancer. Lung Cancer 2011; 74: 481–485.
Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005; 435: 677–681.
van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell 2006; 10: 389–399.
Zhang H, Nimmer PM, Tahir SK, Chen J, Fryer RM, Hahn KR et al. Bcl-2 family proteins are essential for platelet survival. Cell Death Differ 2007; 14: 943–951.
Schoenwaelder SM, Jarman KE, Gardiner EE, Hua M, Qiao J, White MJ et al. Bcl-xL-inhibitory BH3 mimetics can induce a transient thrombocytopathy that undermines the hemostatic function of platelets. Blood 2011; 118: 1663–1674.
Mason KD, Carpinelli MR, Fletcher JI, Collinge JE, Hilton AA, Ellis S et al. Programmed anuclear cell death delimits platelet life span. Cell 2007; 128: 1173–1186.
Wilson WH, O'Connor OA, Czuczman MS, LaCasce AS, Gerecitano JF, Leonard JP et al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: a phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol 2010; 11: 1149–1159.
Gandhi L, Camidge DR, Ribeiro de Oliveira M, Bonomi P, Gandara D, Khaira D et al. Phase I study of Navitoclax (ABT-263), a novel Bcl-2 family inhibitor, in patients with small-cell lung cancer and other solid tumors. J Clin Oncol 2011; 29: 909–916.
Tolcher AW, LoRusso P, Arzt J, Busman TA, Lian G, Rudersdorf NS et al. Safety, efficacy, and pharmacokinetics of navitoclax (ABT-263) in combination with irinotecan: results of an open-label, phase 1 study. Cancer Chemother Pharmacol 2015; 76: 1041–1049.
Kipps TJ, Eradat H, Grosicki S, Catalano J, Cosolo W, Dyagil IS et al. A phase 2 study of the BH3 mimetic BCL2 inhibitor navitoclax (ABT-263) with or without rituximab, in previously untreated B-cell chronic lymphocytic leukemia. Leuk Lymphoma 2015; 56: 2826–2833.
Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med 2013; 19: 202–208.
Roberts AW, Davids MS, Pagel JM, Kahl BS, Puvvada SD, Gerecitano JF et al. Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia. N Engl J Med 2016; 374: 311–322.
Stilgenbauer S, Eichhorst B, Schetelig J, Coutre S, Seymour JF, Munir T et al. Venetoclax in relapsed or refractory chronic lymphocytic leukaemia with 17p deletion: a multicentre, open-label, phase 2 study. Lancet Oncol 2016; 17: 768–778.
Konopleva M, Pollyea DA, Potluri J, Chyla B, Hogdal L, Busman T et al. Efficacy and biological correlates of response in a phase ii study of venetoclax monotherapy in patients with acute myelogenous leukemia. Cancer Discov 2016; 6: 1106–1117.
Pratz K, Pollyea D, Jonas B, Pullarkat V, Wei A, Arellano M et al. Safety and efficacy of venetoclax in combination with decitabine or azacitidine in treatment-naive, elderly patients ≥65 years with acute myeloid leukemia. Eur Hematol Assoc Madrid, Spain 2017.
Sarosiek KA, Letai A . Directly targeting the mitochondrial pathway of apoptosis for cancer therapy using BH3 mimetics - recent successes, current challenges and future promise. FEBS J 2016; 283: 3523–3533.
Wong M, Tan N, Zha J, Peale FV, Yue P, Fairbrother WJ et al. Navitoclax (ABT-263) reduces Bcl-x(L)-mediated chemoresistance in ovarian cancer models. Mol Cancer Ther 2012; 11: 1026–1035.
Heere-Ress E, Thallinger C, Lucas T, Schlagbauer-Wadl H, Wacheck V, Monia BP et al. Bcl-X(L) is a chemoresistance factor in human melanoma cells that can be inhibited by antisense therapy. Int J Cancer 2002; 99: 29–34.
Rudin CM, Hann CL, Garon EB, Ribeiro de Oliveira M, Bonomi PD, Camidge DR et al. Phase II study of single-agent navitoclax (ABT-263) and biomarker correlates in patients with relapsed small cell lung cancer. Clin Cancer Res 2012; 18: 3163–3169.
Zhang H, Guttikonda S, Roberts L, Uziel T, Semizarov D, Elmore SW et al. Mcl-1 is critical for survival in a subgroup of non-small-cell lung cancer cell lines. Oncogene 2011; 30: 1963–1968.
Derenne S, Monia B, Dean NM, Taylor JK, Rapp MJ, Harousseau JL et al. Antisense strategy shows that Mcl-1 rather than Bcl-2 or Bcl-x(L) is an essential survival protein of human myeloma cells. Blood 2002; 100: 194–199.
Sieghart W, Losert D, Strommer S, Cejka D, Schmid K, Rasoul-Rockenschaub S et al. Mcl-1 overexpression in hepatocellular carcinoma: a potential target for antisense therapy. J Hepatol 2006; 44: 151–157.
Glaser SP, Lee EF, Trounson E, Bouillet P, Wei A, Fairlie WD et al. Anti-apoptotic Mcl-1 is essential for the development and sustained growth of acute myeloid leukemia. Genes Dev 2012; 26: 120–125.
Opferman JT, Letai A, Beard C, Sorcinelli MD, Ong CC, Korsmeyer SJ . Development and maintenance of B and T lymphocytes requires anti-apoptotic MCL-1. Nature 2003; 426: 671–676.
Wang X, Bathina M, Lynch J, Koss B, Calabrese C, Frase S et al. Deletion of MCL-1 causes lethal cardiac failure and mitochondrial dysfunction. Genes Dev 2013; 27: 1351–1364.
Perciavalle RM, Stewart DP, Koss B, Lynch J, Milasta S, Bathina M et al. Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nat Cell Biol 2012; 14: 575–583.
Friberg A, Vigil D, Zhao B, Daniels RN, Burke JP, Garcia-Barrantes PM et al. Discovery of potent myeloid cell leukemia 1 (Mcl-1) inhibitors using fragment-based methods and structure-based design. J Med Chem 2013; 56: 15–30.
Stewart ML, Fire E, Keating AE, Walensky LD . The MCL-1 BH3 helix is an exclusive MCL-1 inhibitor and apoptosis sensitizer. Nat Chem Biol 2010; 6: 595–601.
Leverson JD, Zhang H, Chen J, Tahir SK, Phillips DC, Xue J et al. Potent and selective small-molecule MCL-1 inhibitors demonstrate on-target cancer cell killing activity as single agents and in combination with ABT-263 (navitoclax). Cell Death Dis 2015; 6: e1590.
Letai A . S63845, an MCL-1 selective BH3 mimetic: another arrow in our quiver. Cancer Cell 2016; 30: 834–835.
Vogler M, Butterworth M, Majid A, Walewska RJ, Sun XM, Dyer MJ et al. Concurrent up-regulation of BCL-XL and BCL2A1 induces approximately 1000-fold resistance to ABT-737 in chronic lymphocytic leukemia. Blood 2009; 113: 4403–4413.
Morales AA, Kurtoglu M, Matulis SM, Liu J, Siefker D, Gutman DM et al. Distribution of Bim determines Mcl-1 dependence or codependence with Bcl-xL/Bcl-2 in Mcl-1-expressing myeloma cells. Blood 2011; 118: 1329–1339.
Ryan J, Letai A . BH3 profiling in whole cells by fluorimeter or FACS. Methods 2013; 61: 156–164.
Ryan J, Montero J, Rocco J, Letai A . iBH3: simple, fixable BH3 profiling to determine apoptotic priming in primary tissue by flow cytometry. Biol Chem 2016; 397: 671–678.
Ryan JA, Brunelle JK, Letai A . Heightened mitochondrial priming is the basis for apoptotic hypersensitivity of CD4+ CD8+ thymocytes. Proc Natl Acad Sci USA 2010; 107: 12895–12900.
Foight GW, Ryan JA, Gulla SV, Letai A, Keating AE . Designed BH3 peptides with high affinity and specificity for targeting Mcl-1 in cells. ACS Chem Biol 2014; 9: 1962–1968.
Pan R, Hogdal LJ, Benito JM, Bucci D, Han L, Borthakur G et al. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov 2014; 4: 362–375.
Benito JM, Godfrey L, Kojima K, Hogdal L, Wunderlich M, Geng H et al. MLL-rearranged acute lymphoblastic leukemias activate BCL-2 through H3K79 methylation and are sensitive to the BCL-2-specific antagonist ABT-199. Cell Rep 2015; 13: 2715–2727.
Montero J, Stephansky J, Cai T, Griffin GK, Cabal-Hierro L, Togami K et al. Blastic plasmacytoid dendritic cell neoplasm is dependent on BCL2 and sensitive to venetoclax. Cancer Discov 2017; 7: 156–164.
Anderson NM, Harrold I, Mansour MR, Sanda T, McKeown M, Nagykary N et al. BCL2-specific inhibitor ABT-199 synergizes strongly with cytarabine against the early immature LOUCY cell line but not more-differentiated T-ALL cell lines. Leukemia 2014; 28: 1145–1148.
Montero J, Sarosiek KA, DeAngelo JD, Maertens O, Ryan J, Ercan D et al. Drug-induced death signaling strategy rapidly predicts cancer response to chemotherapy. Cell 2015; 160: 977–989.
Etchin J, Montero J, Berezovskaya A, Le BT, Kentsis A, Christie AL et al. Activity of a selective inhibitor of nuclear export, selinexor (KPT-330), against AML-initiating cells engrafted into immunosuppressed NSG mice. Leukemia 2015; 30: 190–199.
Townsend EC, Murakami MA, Christodoulou A, Christie AL, Koster J, DeSouza TA et al. The public repository of xenografts enables discovery and randomized phase II-like trials in mice. Cancer Cell 2016; 30: 183.
Wu SC, Li LS, Kopp N, Montero J, Chapuy B, Yoda A et al. Activity of the type II JAK2 inhibitor CHZ868 in B cell acute lymphoblastic leukemia. Cancer Cell 2015; 28: 29–41.
Cleary JM, Lima CM, Hurwitz HI, Montero AJ, Franklin C, Yang J et al. A phase I clinical trial of navitoclax, a targeted high-affinity Bcl-2 family inhibitor, in combination with gemcitabine in patients with solid tumors. Invest New Drugs 2014; 32: 937–945.
Ju W, Zhang M, Wilson KM, Petrus MN, Bamford RN, Zhang X et al. Augmented efficacy of brentuximab vedotin combined with ruxolitinib and/or Navitoclax in a murine model of human Hodgkin's lymphoma. Proc Natl Acad Sci USA 2016; 113: 1624–1629.
Will B, Siddiqi T, Jorda MA, Shimamura T, Luptakova K, Staber PB et al. Apoptosis induced by JAK2 inhibition is mediated by Bim and enhanced by the BH3 mimetic ABT-737 in JAK2 mutant human erythroid cells. Blood 2010; 115: 2901–2909.
Kim EY, Jung JY, Kim A, Chang YS, Kim SK . ABT-737 synergizes with cisplatin bypassing aberration of apoptotic pathway in non-small Cell lung cancer. Neoplasia 2017; 19: 354–363.
Deng J, Isik E, Fernandes SM, Brown JR, Letai A, Davids MS . Bruton's tyrosine kinase inhibition increases BCL-2 dependence and enhances sensitivity to venetoclax in chronic lymphocytic leukemia. Leukemia 2017; 31: 2075–2084.
Panayotopoulou EG, Muller AK, Borries M, Busch H, Hu G, Lev S . Targeting of apoptotic pathways by SMAC or BH3 mimetics distinctly sensitizes paclitaxel-resistant triple negative breast cancer cells. Oncotarget 2017.
Kuroda J, Puthalakath H, Cragg MS, Kelly PN, Bouillet P, Huang DC et al. Bim and Bad mediate imatinib-induced killing of Bcr/Abl+ leukemic cells, and resistance due to their loss is overcome by a BH3 mimetic. Proc Natl Acad Sci USA 2006; 103: 14907–14912.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by F Pentimalli
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Montero, J., Letai, A. Why do BCL-2 inhibitors work and where should we use them in the clinic?. Cell Death Differ 25, 56–64 (2018). https://doi.org/10.1038/cdd.2017.183
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2017.183
This article is cited by
-
BH3 mimetics and azacitidine show synergistic effects on juvenile myelomonocytic leukemia
Leukemia (2024)
-
Preparation of biogenic silver chloride nanoparticles from microalgae Spirulina Platensis extract: anticancer properties in MDA-MB231 breast cancer cells
Molecular Biology Reports (2024)
-
Bcl-2 pathway inhibition in solid tumors: a review of clinical trials
Clinical and Translational Oncology (2023)
-
High caspase 3 and vulnerability to dual BCL2 family inhibition define ETO2::GLIS2 pediatric leukemia
Leukemia (2023)
-
Transcriptional signatures of the BCL2 family for individualized acute myeloid leukaemia treatment
Genome Medicine (2022)
