
Abstract
Autophagy is a highly complicated process with participation of large numbers of autophagy-related proteins. Under nutrient starvation, autophagy promotes cell survival by breaking down nonessential cellular components for recycling use. However, due to its high complexity, molecular mechanism of autophagy is still not fully understood. In the present study, we report a novel autophagy-related protein TM9SF4, which plays a functional role in the induction phase of autophagic process. TM9SF4 proteins were abundantly expressed in the kidney, especially in renal proximal tubular epithelial cells. At subcellular cells, TM9SF4 proteins were mostly localized in lysosome, Golgi, late endosome and autophagosome. Knockdown of TM9SF4 with TM9SF4-shRNAs markedly reduced the starvation-induced autophagy in HEK293 cells, the effect of which persisted in the presence of bafilomycin A1. TM9SF4-shRNAs also substantially attenuated the starvation-induced mTOR inactivation. In animal model, starvation was able to induce LC3-II accumulation and cause mTOR inactivation in renal cortical tissue in wild-type mice, the effect of which was minimal/absent in TM9SF4 knockout (TM9SF4−/−) mice. Co-immunoprecipitation and proximity ligation assay demonstrated physical interaction of TM9SF4 proteins with mTOR. In addition, knockdown or knockout of TM9SF4 reduced the starvation-induced cell death in HEK293 cells and animal model. Taken together, the present study identifies TM9SF4 as a novel autophagy-related protein. Under nutrient starvation, TM9SF4 functions to facilitate mTOR inactivation, resulting in an enhanced autophagic flux, which serves to protect cells from apoptotic cell death.
Similar content being viewed by others


Main
TM9SF proteins constitute a well-defined family, characterized by the presence of a large variable extracellular domain and nine putative transmembrane domains.1 TM9SF family is highly conserved through evolution, expressed in Dictyostelium discoideum, Saccharomyces cerevisiae, Arabidosis thaliana, Drosophila melanogaster and mammals.2, 3, 4, 5 TM9SF4 is the fourth member of TM9SF family. The high degree of evolutionary conservation suggests that TM9SF proteins may play an important role in cellular physiology.
Literature shows that TM9SF4 plays important roles in diverse cellular processes. In Dictyostelium amoebae and Drosophila melanogaster, TM9SF4 homologs are found to be involved in cell adhesion, phagocytosis and innate immunity.6, 7, 8, 9, 10 In mammalian cells, TM9SF4 plays a role in leukemic cell adhesion11 and cannibalistic activity of metastatic melanoma cells.12, 13 Through cannibalistic activity, tumor cells feed upon neighboring cells, which is a crucial survival strategy for the tumors under poor nutrition condition.14 A recent study suggests that TM9SF4 could serve as intramembrane cargo receptors controlling vesicular trafficking, exocytosis and surface localization of glycine-rich membrane proteins.15
Autophagy is a highly conserved process essential for cell survival under stress conditions including starvation, hypoxia and intracellular stress.16 In cancer cells, autophagy is also an adaptive survival mechanism to overcome drug/acidic stress-induced cellular stress and cytotoxicity, including alteration of pH homeostasis.17, 18 Under nutrient starvation, autophagy promotes survival by breaking down nonessential cellular components for recycling use.19 There are three critical control points in autophagy, namely induction, substrate targeting and degradation,19 each of which involves participation of large numbers of autophagy-related proteins/factors.19, 20 Mammalian target of rapamycin (mTOR) is a key regulator in the induction phase of autophagy.19, 21 mTOR associates with several proteins such as Raptor and Tti1 to form mTOR complex 1 (mTORC1). Under nutrition-rich condition, mTORC1 is translocated to lysosome, where mTORC1 is activated by Rheb.21 Activation of mTORC1 inhibits ULK1, thus prevents autophagy initiation. Under nutrient starvation, mTORC1 is released from lysosome, causing it to become cytosolic and inactive, resulting in an increased autophagic flux.21 Therefore, mTORC1 plays a crucial role in nutrient-sensing. Autophagy has been a hot research topic in the last decade with bulks of autophagy-regulating genes being identified. However, due to its high complexity, molecular mechanism of autophagy is still not fully understood.
In the present study, we explored potential function of TM9SF4 proteins in autophagy. The results demonstrated an important role of TM9SF4 in promoting autophagy under starvation in HEK293 cells and mouse renal cortical tissue. The action of TM9SF4 in autophagy is likely mediated through mTOR. During nutrient starvation, TM9SF4 negatively regulates mTOR activity, thereby promotes autophagic flux.
Results
Tissue distribution and subcellular localization of TM9SF4
A polyclonal anti-TM9SF4 antibody was generated, targeting against N-terminal region (145aa–158aa) of TM9SF4. The antibody recognized a main band with molecular weight of ~60 kDa in HEK293 cells (Supplementary Figure S1A). Overexpression of TM9SF4 in HEK293 cells markedly increased the intensity of this band, confirming the specificity of this antibody (Supplementary Figure S1A). In another series of experiments, HEK293 cells were transfected with a V5-tagged TM9SF4 construct. In immunoblots, the band of V5-tagged TM9SF4 with the molecular size of ~60 kDa was recognized by both anti-V5 antibody and anti-TM9SF4 antibody (Supplementary Figure S1B), confirming that the ~60 kDa band belonged to TM9SF4 and that the antibody was of good quality.
Tissue distribution of TM9SF4 proteins was examined by immunoblots. The results showed that TM9SF4 proteins were abundantly expressed in mouse and rat kidney (Figure 1a and Supplementary Figure S1D). Immunostaining of renal tissue sections showed that TM9SF4 proteins were abundantly expressed in mouse and rat renal cortical proximal tubular epithelial cells (Figure 1b and Supplementary Figure S1C). In contrast, TM9SF4 immunoreactivity was absent in renal proximal tubular epithelial cells of TM9SF4−/− mice (Figure 1b), which further supported the specificity of our anti-TM9SF4 antibody. We also examined the expression of TM9SF4 in a panel of different human cell lines. The expression of TM4SF4 proteins was detected in human kidney cell line HK2, leukemia T lymphocyte line Jurkat, ovarian cancer line A2780, pancreatic cell line DU145 and gastric cancer line TMK-1 (Supplementary Figure S1E).

Tissue distribution and subcellular localization of TM9SF4 proteins. (a) Representative immunoblot images (top) and summary data (bottom) showing the expression of TM9SF4 proteins in mouse tissues. GAPDH was used as the house-keeping control gene. Summary data are presented as mean±S.E.M. (n=3). (b) Representative images of the renal cortex of wild-type (WT) and TM9SF4−/− (KO) mice, immunostained with preimmune IgG or anti-TM9SF4 antibody (× 400 magnifications). Brown color represents TM9SF4 signal, while blue color shows cell nuclei from hematoxylin counterstain. n=3. (c and d) Co-localization of TM9SF4 proteins with lysosomal marker lysosome-RFP (c) and autophagosome marker RFP-LC3 (d). Dashed lines in the left panels of (c and d) outline the cell boundary, which could be observed at high magnification in differential interference contrast mode. The right most panels in (c) and (d) were the magnified images of Merge. Scale bar=10 μm. Representatives from five experiments
Subcellular localization of TM9SF4 in subcellullar organelles was determined in HEK293 cells. TM9SF4 showed partial co-localization with lysosome marker Lysosome-RFP (Figure 1c) and autophagosome marker RFP-LC3 (Figure 1d). Quantification of pixel co-localization showed 68±12% (n=5) of RFP-Lysosome co-localized with TM9SF4, whereas 38±3% (n=4) of RFP-LC3 co-localized with TM4SF4. TM9SF4 also had partial co-localization with Golgi marker Giantin C20 (Supplementary Figure S2A), late endosome marker M-6-P receptors (Supplementary Figure S2B) and RAB7 (Supplementary Figure S2C), and endoplasmic reticulum marker KDEL (Supplementary Figure S2D). However, TM9SF4 almost had little or no co-localization with early endosomal marker EEA1 (Supplementary Figure S2E) and mitochondrial marker COX1 (Supplementary Figure S2F).
TM9SF4-shRNAs reduced the starvation-stimulated autophagic flux in HEK293 cells
HEK293 cells were exposed to amino acid deprivation in Earle’s Balanced Salt Solution (EBSS) for different time durations to stimulate autophagic flux. As expected, amino acid starvation caused an accumulation of microtubule-associated protein 1 light chain 3-II (LC3-II) (Figures 2a and b), which is a commonly used index for autophagy.19, 22 Starvation also increased the expression level of TM9SF4 proteins (Figures 2a and c).

Knockdown of TM9SF4 using lenti-TM9SF4-shRNA1 inhibited the starvation-induced autophagic flux in HEK293 cells. (a–c) Amino acid starvation in EBSS for 4 and 6 h increased the expression of TM9SF4 and LC3-II in HEK293 cells. Shown are representative immunoblot images (a) and data summary (b and c). (d and e) Representative immunoblot images (d) and data summary (e) demonstrating the effectiveness of TM9SF4-shRNA1 in knocking-down TM9SF4 expression in HEK293 cells under basal and starvation conditions. Lenti-TM9SF4-shRNA1 or its scrambled control was stably transduced into HEK293. (f and g) Effect of TM9SF4-shRNA1 in reducing the LC3-II level in the presence or absence of bafilomycin A1 (Baf, 10 nM). Shown are representative immunoblot images (f) and data summary (g). (h and i) Effect of TM9SF4-shRNA1 in reducing GFP-LC3 puncta formation in the presence or absence of bafilomycin A1 (Baf, 10 nM). Shown are LC3 fluorescent signals from representative single cells (h) and data summary (i). (j and k) HEK293 cells were transfected with a tandem reporter RFP-GFP-LC3. Shown are the effect of TM9SF4-shRNA1 on the formation of autophagosome (yellow dots in merged images) and autolysosome (red dots in merged images) with representative images (j) and data summary (k). In (f–k), amino acid starvation was carried out in EBSS for 2 h. In (h–k), quantification of autophagosome and autolysosome were performed using ImageJ. Scale bar=10 μm. Summary data are presented as mean±S.E.M. (n as labeled in the figures; >100 cells per experiment in (i and k). *P<0.05; **P<0.01. The values in (b,c,e and g) were normalized to β-actin level
A lentiviral-based TM9SF4-shRNA (lenti-TM9SF4-shRNA1) was constructed. The lenti-TM9SF4-shRNA1 could effectively knockdown TM9SF4 proteins by 85% and 68% for cells under basal and amino acid starvation condition, respectively (Figures 2d and e). Importantly, stable expression of lenti-TM9SF4-shRNA1 reduced the LC3-II accumulation under amino acid starvation (Figures 2f and g). A reduced LC3-II could be explained by a decrease in autophagic induction or an increase in autolysosomal degradation. We used bafilomycin A1 to inhibit autolysosomal degradation. In the presence of 10 nM bafilomycin A1, lenti-TM9SF4-shRNA1 still markedly reduced the LC3-II accumulation under starvation (Figures 2f and g). These results were confirmed using another lenti-TM9SF4-shRNA2 (Supplementary Figures S3A–C). These data suggest that TM9SF4 promoted autophagic flux mainly through its action on autophagic induction.
GFP-LC3 puncta formation was quantified under a confocal microscope, which was used as another index of autophagy.22 In the control cells transfected with lenti-scrambled-shRNA, starvation increased the number of GFP-LC3 puncta per cell (Figures 2h and i). Ten nanomolar bafilomycin A1 further increased the GFP-LC3 puncta formation (Figures 2h and i). Importantly, lenti-TM9SF4-shRNA1 markedly inhibited the starvation-induced GFP-LC3 puncta formation (Figures 2h and i). The effect of TM9SF4-shRNA1 on GFP-LC3 puncta formation persisted in the presence of 10 nM bafilomycin A1 (Figures 2h and i).
We then transfected HEK293 cells with the tandem reporter RFP-GFP-LC3, which can effectively and conveniently monitor autophagic flux.23 In this assay, autophagosomes are labeled with dual red and green fluorescence and autolysosomes with red only.23 The results confirmed that amino acid starvation increased the number of autophagosome (yellow dots in merged pictures; Figures 2j and k) and autolysosome (red dots in merged pictures; Figures 2j and k). TM9SF4-shRNA1 reduced the formation of autophagosome and autolysosome under starvation (Figures 2j and k).
Note that, although knockdown of TM9SF4 with lenti-TM9SF4-shRNAs suppressed the autophagic flux under amino acid starvation condition, it had little or no effect on autophagic flux in cells under basal non-starvation condition (Figures 2f–k).
Overexpression of TM9SF4 promoted the autophagic flux
The effect of TM9SF4 overexpression on autophagy was examined (Figure 3). TM9SF4 overexpression promoted the autophagic flux as indicated by a small but statistically significant increase in the LC3-II level under both basal and amino acid starvation conditions (Figures 3a and b). This effect persisted in the presence of 10 nM bafilomycin A1 at least at basal non-starvation condition (Figures 3a and b). TM9SF4 overexpression also increased LC3 puncta formation (RFP-LC3), the effect of which persisted in the presence of 10 nM bafilomycin A1 (Figures 3c and d).

Overexpression of TM9SF4 promoted autophagic flux in HEK293 cells. (a and b) Effect of TM9SF4 overexpression on LC3-II level in the presence or absence of bafilomycin A1 (Baf, 10 nM). Shown are representative immunoblot images (a) and data summary (b). (c and d) Effect of TM9SF4 overexpression on RFP-LC3 puncta formation. Shown are LC3 fluorescent signals from representative single cells (c) and data summary (d). The right most panels in (c) were the magnified images of Merge. Scale bar=10 μm. The quantification of RFP-LC3-positive autophagosome was performed using ImageJ. Amino acid starvation was carried out in EBSS for 2 h. Summary data are presented as mean±S.E.M. (n=6 in B; n=5 and >100 cells per experiment in (d). The values in (b) were normalized to β-actin level. *P<0.05; **P<0.01
TM9SF4 inhibited mTOR activity
We examined the possible role of TM9SF4 in regulating mTOR activity. In control cells expressing lenti-scrambled-shRNA, amino acid starvation for 4 and 6 h reduced mTOR activity, as indicated by a decreased level of phosphorylated mTOR (Figures 4a and b). Eukaryotic translation initiation factor 4E–binding protein 1 (4E-BP1) and ULK1 are downstream phosphorylation substrates of mTOR.24 Amino acid starvation also decreased the level of phosphorylated 4E-BP1 (Figures 4a and c) and ULK1 (Figures 4a and d). Intriguingly, suppression of TM9SF4 by stable expression of each of the two lenti-TM9SF4-shRNAs substantially attenuated the starvation-stimulated dephosphorylation of mTOR, 4E-BP1 (Figures 4a–d and Supplementary Figures S3D–F). Lenti-TM9SF4-shRNA1 also attenuated the starvation-stimulated dephosphorylation of ULK1 (Figures 4a and d). However, under basal non-starvation condition, lenti-TM9SF4-shRNAs had little or no effect on the phosphorylation of mTOR, 4E-BP1 and ULK1 (Figures 4a–d). These data suggest that TM9SF4 functions to promote mTOR dephosphorylation (or inactivate mTOR) under starvation condition. In addition, we also used rapamycin to inactivate mTOR (Supplementary Figure S4). Suppression of TM9SF4 by lenti-TM9SF4-shRNA1 also attenuated the rapamycin-induced dephosphorylation of mTOR (Supplementary Figure S4).

TM9SF4 inhibited mTOR activity in HEK293 cells. (a–d) Representative immunoblot images (a) and data summary (b–d) showing the effect of TM9SF4-shRNA1 on the expressional levels of phospho-mTOR/mTOR, phospho-4E-BP1/4E-BP1 and phospho-ULK1-S757/ULK1. (e–h) Representative immunoblot images (e) and data summary (f–h) showing the effect of TM9SF4 overexpression on the expressional levels of phospho-mTOR/mTOR, phospho-4E-BP1/4E-BP1 and phospho-ULK1-S757/ULK1. (i and j) Representative immunoblot images (i) and data summary (j) showing that mTOR-siRNA caused LC3-II re-accumulation in lenti-TM9SF4-shRNA1-expressing cells. mTOR-siRNA could effectively knockdown the expression level of mTOR proteins (i and k). Amino acid starvation was carried out in EBSS for 4 and 6 h in (a–d), 4 h in (e–k). Summary data are presented as mean±S.E.M. (n as labeled in the figures). The values in summary data were normalized to total protein levels of mTOR, 4E-BP1 and β-actin, respectively. *P<0.05; **P<0.01
Conversely, TM9SF4 overexpression had the opposite effect. As shown in Figures 4e–h, TM9SF4 overexpression promoted dephosphorylation of mTOR, 4-EBP1 and ULK1 under both basal and starvation conditions.
To further confirm whether TM9SF4 indeed acted through mTOR to modulate autophagic process, we attempted to rescue TM9SF4 silencing by co-silencing mTOR. As expected, stable expression of lenti-TM9SF4-shRNA1 reduced LC3-II accumulation under amino acid starvation in HEK293 cells (Figures 4i and j). These lenti-TM9SF4-shRNA1-expressing HEK293 cells were further transfected with mTOR-siRNA or scrambled-siRNA (mTOR-scr). The results showed that mTOR-siRNA reversed the effect of TM9SF4 silencing, causing re-accumulation of LC3-II (Figures 4i and j). As a control, the mTOR-siRNA could effectively knock down the expression of mTOR (Figures 4i and k). These data further support the notion that TM9SF4 acts through mTOR to regulate autophagy.
Physical association of TM9SF4 with mTOR
mTOR proteins interact with several binding partners to form physical complexes.24 Thus we examined the possibility of whether mTOR could physically interact with TM9SF4 proteins. In co-immunoprecipitation experiments, anti-TM9SF4 antibody could pull down mTOR proteins in both control and starved cells (Figure 5a). Furthermore, anti-mTOR antibody could reciprocally pull-down TM9SF4 proteins (Figure 5b). As control, the pull-down experiments were also performed using preimmune IgG as the pulling antibody. As expected, no band was observed in these negative control experiments (Figures 5a and b).

Physical interaction of TM9SF4 with mTOR in HEK293 cells. (a and b) Representative immunoblot images of co-immunoprecipitation experiments. The pulling antibodies (anti-TM9SF4 antibody in a and anti-mTOR antibody in b) and the blotting antibodies were indicated. Control immunoprecipitation was performed using preimmune IgG (labeled as pre-IgG). The cell lysates were displayed as input. n=5. (c and d) Representative images (c) of TM9SF4/mTOR interaction together quantification (d) of fluorescent spots using proximity ligation assay (PLA). Summary data are presented as mean ± S.E.M. (n=4 experiments). **P<0.01. Scale bar=20 μm
Proximity ligation assay (PLA) allows the detection of protein interaction in situ in unmodified cells, in which each interaction site is visualized as a distinct fluorescent punctum.25 The PLA results demonstrated a large number of distinct fluorescent puncta, representing interaction sites of TM9SF4 with mTOR in HEK293 cells under basal non-starved condition (Figures 5c and d). Interestingly, the interaction puncta greatly reduced under starvation condition (Figures 5c and d).
Subcellular immunolocalization also demonstrated a partial co-localization of TM9SF4 with mTOR in non-starved control cells (Supplementary Figure S5A). The co-localization reduced in starved cells (Supplementary Figure S5A). Quantification of pixel co-localization showed 46±4% (n=4) of mTOR co-localized with TM9SF4 under non-starved condition, while the co-localization reduced to 30±1% (n=4) under starvation. mTOR localization in lysosome was also confirmed, the degree of which decreased under amino acid starvation (Supplementary Figure S5B), supporting the notion of mTOR dissociation from lysosome under starvation.21 Three-dimensional (3D) confocal microscopy fluorescent images were used to better visualize the co-localization of TM9SF4 with mTOR, an mTOR complex 1 protein Raptor, and an mTOR complex 2 protein Rictor.24 Partial co-localization of TM9SF4 with mTOR and Raptor was observed (Supplementary Figure S6). The degree of co-localization decreased under amino acid starvation (Supplementary Figure S6). mTOR did not show significant co-localization with Rictor under both basal and starvation conditions (Supplementary Figure S6).
Knockdown of TM9SF4 aggravated starvation-induced cell death
We explored the possible role of TM9SF4 in starvation-induced cell death. MTT assay showed that the amino acid starvation caused a reduction in cell metabolic activity in a time-dependent manner in cells expressing either lenti-scrambled-shRNA or lenti-TM9SF4-shRNA1 (Figure 6a). However, compared with the control cells expressing lenti-scrambled-shRNA, the cells expressing lenti-TM9SF4-shRNA1 had lower cell viability in MTT assay.

TM9SF4-shRNA1 aggravated the starvation-induced cell death in HEK293 cells. HEK293 cells were stably transfected with lenti-scrambled-shRNA or lenti-TM9SF4-shRNA1. Amino acid starvation was carried out in EBSS for 2−6 h in (a), or 1−3 h in (b and c). (a) Cell viability assay by MTT. The data are expressed as the percent of metabolic activity relative to non-starved cells. (b and c) Cell death assay based on Apopxin green indicator and 7-amino-actinomycin (7-AAD) using Flow Cytometry. Apopxin green(−)/7-AAD (+) cells (Q1) were considered necrotic; Apopxin green(+)/7-AAD(+) double-positive cells (Q2) were considered late apoptotic; Apopxin green(−)/7-AAD(−) cells (Q3) were considered alive; and Apopxin green(+)/7-AAD(−) cells (Q4) were considered early apoptotic. Quantitative analysis of the apoptotic (Q2+Q4) cells by fluorescence-activated cell sorting (FACS) analysis is summarized in (c). Summary data are presented as mean±S.E.M. (n=7 in a; n=4 in c). *P<0.05
Flow cytometric analysis was used to distinguish early apoptotic, late apoptotic and necrotic cells. In control cells expressing lenti-scrambled-shRNA, 1 and 3 h starvation markedly increased the population of Apopxin Green(+)/7-AAD(−) cells, which represented early apoptotic cells. Expression of lenti-TM9SF4-shRNA1 further increased the number of total apoptotic cells (Apopxin Green (+)/7-AAD(−) plus Apopxin Green (+)/7-AAD(+)) under starvation condition (Figures 6b and c).
We also examined the effect of TM9SF4 overexpression on cell death by MTT assay. However, TM9SF4 overexpression failed to protect HEK293 cells from starvation-induced cell death in MTT assay (Supplementary Figure S7).
Role of TM9SF4 in autophagy and apoptosis in mouse kidney in vivo
TM9SF4 function in autophagy was further studied in mouse kidney in vi vo using TM9SF4−/− mice. The genotype of TM9SF4−/− mice was verified by tail DNA genotyping (Supplementary Figure S8). No apparent gross abnormality was observed in TM9SF4−/− mice under normal feeding condition. But database (http://www.informatics.jax.org/allele/allgenoviews/MGI:4363779) reported that these mice have abnormality in some skeleton bones, blood cholesterol and circulating Ca2+.
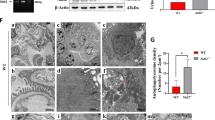
Animals were subjected to food starvation for 24 h, only supplied with drinking water, with or without intraperitoneal injection of bafilomycin A1 (25 ng/g body weight). In wild-type mice, the starvation caused a large increase in LC3-II level in the renal cortex, which became even more marked in the presence of bafilomycin A1 (Figures 7a and b). Intriguingly, this starvation-induced LC3-II elevation in the renal cortex became minimal in TM9SF4−/− mice (Figures 7a and b). In addition, 24 h starvation also reduced the level of phosphorylated mTOR and 4-EBP1 in the renal cortex of wild-type mice (Figures 7c–e). Again, this starvation effect on the phosphorylation levels of mTOR and 4-EBP1 diminished in TM9SF4 −/− mice (Figures 7c–e).

TM9SF4 promoted autophagy in mouse renal tissues in vivo. (a and b) Representative immunoblots (a) and data summary (b) showing LC3-II protein level in the renal cortex of wild-type (WT) and TM9SF4−/− (KO) mice. (c–e) Representative immunoblots (c) and data summary (d and e) showing the protein levels of phospho-mTOR (d) and phosphor`E-BP1 (e) in the renal cortex of wild-type and TM9SF4−/− mice. (f and g) Representative pictures (f) and summary data (g) of TUNEL-positive cells in renal cortical tissue sections prepared from wild-type and TM9SF4−/− mice. The nuclei were stained blue with DAPI. Green signal indicate apoptotic nuclei. Animals were starved for 24 h with or without bafilomycin A1 (Baf, 25 ng/g body weight). Control had no starvation. Summary data are presented as mean±S.E.M. (n=5 in B; n=4 in (d,e and g). The values in (b) were normalized to β-actin, and those in (d and e) were normalized to total protein levels of mTOR and 4E-BP1, respectively. *P<0.05; **P<0.01. Scale bar=20 μm
We also studied the role of TM9SF4 in apoptotic cell death in mouse renal cortex using terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay. The results showed that TM9SF4−/− mice has an increased apoptotic cell death under both basal and starvation condition compared with those of wild-type mice (Figures 7f and g).
Role of TM9SF4 in modulating reactive oxygen species production
Mitochondrial integrity was examined by fluorescent dye Mitotracker Red, whose uptake depends on mitochondrial membrane potential.26, 27 In agreement with other reports,28 starvation increased the mitochondrial membrane potentials (Supplementary Figure S9). However, TM9SF4 knockdown or overexpression did not cause mitochondrial damage in HEK293 cells, as indicated by no significant change in mitochondrial membrane potentials (Supplementary Figure S9).
Reactive oxygen species (ROS) production was monitored by dihydroethidium (DHE) dye. Compared with lenti-scrambled-shRNA, lenti-TM9SF4-shRNA1 treatment increased the production of ROS under starvation, as indicated by increased DHE fluorescence intensity (Supplementary Figure S10). DHE staining was also performed in cryo-sectioned renal cortical tissue samples. TM9SF4−/− mice displayed an elevated ROS production under both basal and starvation condition compared with those of wild-type mice (Supplementary Figure S10), suggesting that TM9SF4 serves to reduce ROS production.
Discussion
The main findings of the present study are as follows: (1) TM9SF4 proteins were found to be abundantly expressed in rodent kidney. At subcellular cells, TM9SF4 proteins were localized in lysosome, Golgi, late endosome and autophagosome. (2) Knockdown of TM9SF4 with TM9SF4-shRNAs markedly reduced the starvation-induced autophagic flux in HEK293 cells. The effect of TM9SF4-shRNAs persisted in the presence of bafilomycin A1. (3) Knockdown of TM9SF4 with TM9SF4-shRNAs substantially attenuated the starvation-induced mTOR inactivation. Co-immunoprecipitation, PLA and subcellular co-localization experiments suggested physical association of TM9SF4 with mTOR. (4) In animal models, TM9SF4 was found to be abundantly expressed in renal proximal tubules in wild-type mice but not in TM9SF4−/− mice. Starvation was able to induce LC3-II accumulation and mTOR inactivation in renal cortical tissue in wild-type mice, the effect of which was minimal/absent in TM9SF4−/− mice. (5) Knockdown or knockout of TM9SF4-shRNA aggravated the starvation-induced apoptotic cell death in HEK293 cells and animal model. Taken together, the present study identifies TM9SF4 as a novel autophagy-related protein that is critically involved in mTOR inactivation under nutrient starvation, and that TM9SF4 functions to protect cells from apoptotic cell death.
We first explored the functional role of TM9SF4 in starvation-induced autophagy using HEK293 cell line. Nutrient starvation stimulated autophagic flux, as indicated by LC3-II accumulation in immunoblots, an increase in the formation of GFP-LC3 puncta, and simultaneous accumulation of autophagosome and autolysosome in RFP-GFP-LC3 tandem reporter assay. Importantly, knockdown of TM9SF4 with TM9SF4-shRNA markedly reduced these autophagy-related indexes. TM9SF4-shRNA also aggravated the starvation-induced apoptotic cell death, as demonstrated in MTT assay and flow cytometric analysis of Apopxin Green/7-AAD. These data suggest a critical role of TM9SF4 in promoting autophagic flux, which exerted a cytoprotective role against cell death under nutrient starvation. However, we also found that overexpression of TM9SF4 failed to protect cell from starvation-induced cell death in MTT assay. One possible explanation for this discrepancy is that endogenous TM9SF4 expression was already high in HEK293 cells under starvation, further overexpression may only have little impact on its function.
Autophagy is a highly complicated process including three key phases, namely induction, substrate targeting and degradation.19 An intriguing next question is in which autophagic stage TM9SF4 plays its role, in induction phase or in degradation phase. A recent study showed that TM9SF4 may interact with vacuolar type H+-ATPase, facilitating acidification in lysosome and intracellular vesicles,12, 13 which is important for cargo degradation inside of autolysosomes.22, 29 Thus we used bafilomycin A1 to inhibit vacuolar type H+-ATPase, thereby suppresses autophagosomal degradation. However, in the presence of bafilomycin A1, TM9SF4-shRNA could still markedly attenuate the starvation-induced LC3-II accumulation in immunoblots and GFP-LC3 puncta formation under a fluorescence microscope. In addition, RFP-GFP-LC3 tandem reporter assay showed that TM9SF4 knockdown reduced the formation of both autophagosome and autolysosome. Together, these data suggest that TM9SF4 promotes autophagy mainly through facilitating the induction step of autophagic flux.
We then turned our attention to the induction phase of autophagy. It is well recognized that mTOR could serve as a nutrient sensor.21 Under nutrient starvation, mTOR is released from lysosome, making mTOR to become cytosolic and inactive, consequently enhancing autophagic flux.21 Indeed, we found that amino acid starvation markedly reduced the mTOR activity in HEK293 cells, as indicated by dephosphorylation of mTOR, 4E-BP1 and ULK1 (Figure 4). Intriguingly, TM9SF4-shRNAs substantially attenuated the starvation-induced mTOR inactivation, as indicated by phosphorylation level of mTOR, 4E-BP1 and ULK1. These data provide strong evidence that TM9SF4 acts through mTOR to facilitate autophagic flux. Interestingly, proximity ligation assay and co-immunoprecipitation experiments both demonstrated that TM9SF4 physically associates with mTOR. Subcellular co-localization studies also confirmed partial co-localizations of TM9SF4 with mTOR and its binding partner Raptor. Together, these data suggest that TM9SF4 interacts with mTOR to reduce its activity under starvation.
Next intriguing question is how TM9SF4 could inactivate mTOR under starvation. In this regard, both proximity ligation assay (Figures 5c and d) and subcellular immune-co-localization experiments (Supplementary Figures S5A and S6) clearly demonstrated that amino acid starvation reduced the physical interaction of TM9SF4 with mTOR. Based on this, we speculated that TM9SF4, which is partially located in lysosome (Figure 1c) and is known to modulate vesicular trafficking,15 may promote mTOR dissociation from lysosome, causing its inactivation. However, this is highly speculative. Further study is needed to clarify the underlying mechanism of how TM9SF4 could inactivate mTOR under nutrition starvation.
Kidney tubular epithelial cells, especially proximal tubular cells, consume a large amount of energy in the process of substance reabsorption.30 In addition, renal proximal tubules are susceptible to various kinds of insults such as ischemia–reperfusion injury and nephrotoxic substrates.30, 31, 32 Because the proximal tubules are energy wasting and susceptible to injury, autophagy plays an important role in the physiology and pathology of the proximal tubules. Therefore, we extended our study to mouse renal tubular epithelial cells in vivo. Our results showed that nutrient starvation caused LC3-II accumulation and mTOR inactivation in renal cortical tissue in wild-type mice. But this starvation effect on LC3-II and mTOR was minimal/absent in TM9SF4−/− mice. TUNEL assay demonstrated a protective role of TM9SF4 in renal tissue under starvation. These data agreed with those collected from HEK293 cells, suggesting that TM9SF4 promotes autophagy in kidney, which has a protective role against renal cell death under starvation condition.
We next examined whether TM9SF4 could affect several other autophagy-related pathways including mitochondrial damage and ROS production. TM9SF4 knockdown/knockout was found to have no effect on mitochondrial integrity (Supplementary Figure S9), but it markedly increased ROS production in both the cultured HEK293 cells and renal cortical tissue (Supplementary Figure S10). It is unclear how TM9SF4 knockdown/knockout could result in an increased ROS production. However, it is generally accepted that an impaired autophagy would lead to increase in ROS production.33 Therefore, it is possible that the increased ROS production could result from an impaired autophagic flux due to TM9SF4 knockdown/knockout. Further study is needed to clarify whether this is indeed the case. Taken together, our data suggest that TM9SF4 not only acts through mTOR to promote autophagic flux, but also reduces ROS production, both of which could have cytoprotective roles.
Interestingly, while the present study is focused on the role of TM9SF4 in autophagy or 'self-eating' in normal cells and tissues, previous studies have demonstrated that TM9SF4 is essential for cannibalistic behavior of metastatic melanoma cells.2, 13 Through cannibalistic activity, tumor cells feed upon neighboring cells or debris rather than its internal substances as its survival strategy.14 Furthermore, it has also suggested that TM9SF4, through its interaction with H+-vacuolar-V-ATPase, plays an important role for setting up acidic microenvironment,12 which may in turn promote autophagy and serve to protect tumor cells.17, 18 These previous data provide an exciting possibility of targeting TM9SF4 as a potential anti-cancer strategy.2, 34 Together with the results from the present study, it is clear now that TM9SF4 has cytoprotective function in both metastatic tumor cells and normal cells/tissues under starvation condition.
In conclusion, the present study identified TM9SF4 as a novel autophagy-related protein. Under nutrient starvation, TM9SF4 functions to facilitate mTOR inactivation, resulting in an enhanced autophagic flux. In renal tissue, where TM9SF4 is abundantly expressed, TM9SF4 plays an important role in promoting autophagic flux under nutrient starvation.
Materials and methods
TM9SF4 antibody generation
Rabbit TM9SF4 polyclonal antibodies were generated by immunizing rabbits with the peptide KLH-CELYSNRDSDDKKKE corresponding to a region near the N-terminus of human TM9SF4 (NP_055557.2) (amino acids 145–158), followed by affinity purification and validation by immunoblot analysis.
Construction of plasmids
Human TM9SF4 cDNA (NP_055557.2) was cloned into two vectors: pcDNA6.0 and pEGFP-N1 respectively. TM9SF4-pcDNA6.0 was constructed by inserting TM9SF4 with stop codon into pcDNA6.0 vector at enzymatic sites EcoRI and XhoI, followed by IRES-GFP. TM9SF4-V5 was constructed by inserting the TM9SF4 without stop codon into pcDNA6.0 so that TM9SF4 and V5 were translated continuously. TM9SF4-GFP was constructed by inserting TM9SF4 without stop codon into pEGFP-N1 at enzymatic sites XhoI and BamHI so that TM9SF4 and GFP were translated continuously. RFP-TM9SF4 was constructed in the vector pCMV6-AV-RFP at enzyme sites of SgfI and NotI. pEGFP-LC3 (human) plasmid was purchased from Addgene (ID: 24920, Cambridge, MA, USA). pCMV6-AV-RFP-LC3 was a gift from Dr. Shi TP in Chinese National Human Genome Center, Beijing. LC3-II was inserted in the vector pCMV6-AV-RFP at enzyme sites of SgfI and MluI. RFP-GFP-LC3, a kind gift from Prof. YUE J in the City University of Hong Kong, was inserted in the vector plenti-cmv-dest-puro. All constructs were confirmed by restriction enzyme digestion and DNA sequencing.
Lentiviral packaging of TM9SF4-shRNAs and infection
Short hairpin RNA (shRNA) constructs against human TM9SF4 was generated using pLKO.1 puro vector containing a cassette for puromycin according to the manufacturer’s instructions (OligoEngine, Seattle, WA, USA). The targeted sequence 1 (shRNA1) was 5′-GCGGATCACAGAAGACTACTA-3′. The targeted sequence 2 (shRNA2) was 5′-CGGTGGTACATGAACCGATTT-3′. Scrambled-shRNA was used as control. Lentiviral production was performed by co-transfecting pLKO.1 and packaging vectors into HEK293FT cells. The viruses were purified by centrifuging for 2 h at 50 000 g and then re-suspended in PBS. Lenti-TM9SF4-shRNA and its scrambled control were stably transduced into HEK293 cells with puromycin selection. Knockdown of TM9SF4 was confirmed by RT-PCR and immunoblots.
Cell culture, plasmid and siRNA transfection
HEK293 cells were cultured in Dulbecco’s modified Eagle medium (Invitrogen, Carlsbad, CA, USA) containing 10% fetal bovine serum (Invitrogen) and a combination of penicillin–streptomycin (Invitrogen) at 37 °C in a 5% CO2 atmosphere. Transient transfection of TM9SF4-pcDNA6.0, TM9SF4-GFP, GFP-LC3, pEGFP-N1, RFP-LC3 or RFP-GFP-LC3 in HEK293 cells was conducted using lipofectamine 2000 (Invitrogen) according to the manufacturer’s instruction.
siRNA against human mTOR (NM_004958.3) was synthesized (GenePharma, Shanghai, China). The targeted sequence is 5′-AAAGAUCUCAUGGGCUUTT-3′. Scrambled-siRNA was used as control. Transient transfection of mTOR-siRNA was carried out using Lipofectamine RNAiMAX Transfection Reagent (Thermo Fisher Scientific, MA, USA, 13778075).
Immunoblot
Whole-cell lysates or homogenized tissue samples were extracted with protein extraction buffer, which contained in mM: 50 Tris-HCl, 150 NaCl, 50 NaF and 1.5% Nonidet P-40, 0.5% sodium deoxycholate, pH 7.5, with addition of the protease inhibitor cocktail tablets (Roche, USA). Ten micrograms proteins (for cell samples) or 50 μg proteins (for tissue samples) were loaded onto each lane and separated on 12% SDS/PAGE gel after boiled in SDS loading buffer. Proteins were transferred to a PVDF membrane and probed with appropriate primary antibodies overnight at 4 °C in in PBS containing 5% non-fat milk and 0.1% Tween 20. Immunodetection was accomplished with horseradish peroxidase-conjugated secondary antibody (1:5000), followed by ECL Plus Western blotting detection system (Amersham, Buckinghamshire, England). Immunoblots with anti-β-actin antibody were used to confirm that an equal amount of proteins was loaded onto each lane. The band intensity was analyzed by ImageJ (US National Institutes of Health, Rockville, MD, USA). The primary antibodies include: TM9SF4 (home-made, 1 μg/ml), anti-β-actin (1:2000; Santa Cruz Biotech, Dallas, TX, USA, sc-47778), anti-LC3-II/MAP1LC3II (1:1000; Novus, Littleton, CO, USA, NB100-2220), anti-mTOR (1:1000; Cell Signaling Technology, Danvers, MA, USA, 2983s), anti-p-mTOR (Ser 2448) (1:1000; Cell Signaling Technology, 5536P), anti-Raptor (1:1000; Bethyl Laboratories, Montgomery, TX, USA, A300-553A), anti-Rictor (1:1000; Bethyl Laboratories, Montgomery, TX, USA, A300-459A-3), anti-p-4E-BP1 (1:1000; Cell Signaling Technology, 2855P), anti-4E-BP1(1:2000; Cell Signaling Technology, 9452), anti-p-ULK1-757 (1:1000; Cell Signaling Technology, 6888T), anti-ULK1 (1:1000; Cell Signaling Technology, 8054S) and anti-mTOR (1:1000; Abcam,Cambridge, MA, USA, 87540) for PLA experiment.
For co-immunoprecipitation, whole-cell lysates were extracted with KCl–IP buffer containing 20 mM Tris pH 7.5, 100 mM KCl, 0.5 mM DTT, 0.2% NP-40, 10% glycerol, supplemented with protease inhibitors. TM9SF4 and mTOR proteins were immunoprecipitated by incubating 800 μg of the extracted proteins with anti-TM9SF4 antibody or anti-mTOR antibody, respectively, at 4 °C with rotation for 2 h. Protein G agarose was then added and incubated overnight at 4 °C with rotation. The immunoprecipitates were washed three times with PBS, followed by immunoblotting.
Immunostaining
Double immunolabeling was used for subcellular localization of TM9SF4. Briefly, the cells grown on coverslips were fixed by 4% paraformaldehyde in PBS, permeabilized with 0.4% Triton X-100, following by incubation with primary antibodies at 4 °C overnight. After wash in PBS, the cells were incubated with secondary antibodies for 1 h at room temperature. Lysosome marker is CellLight Lysosomes-RFP, BacMam 2.0 (C10597; Thermo Fisher). Primary antibodies were anti-COX-I (1D6) (1:500; Santa Cruz Biotech, sc-58347), anti-EEA1 antibody (1G11) (1:500; Abcam, ab70521), anti-M-6-P (mannose-6-phosphate receptor) (2G11) (1:500; Abcam, ab2733), anti-RAB7 (1:500; Abcam, ab50533) and anti-Giantin (c-20) (1:50; Santa Cruz Biotech, sc-46991). Secondary antibodies were Alexa Fluor 488 donkey anti-rabbit IgG (H+L) (Abcam, A21206) and Alexa Fluor 555 donkey anti-mouse IgG (H+L) (Abcam, A31570). After washing and mounting, immunofluorescence of the cells was detected using the FV1000 confocal system. For GFP or Alexa Fluor 488, the excitation wavelength was 488 nm. For RFP or Alexa Fluor 555, the excitation wavelength was 543 nm.
For immunostaining of tissue sections, tissues were post-fixed with 4% paraformaldehyde overnight, subsequently embedded with paraffin, and sectioned to 10 μm thicknesses. The sections were dewaxed and rehydrated, followed by overnight incubation at 4 °C with anti-TM9SF4 antibody (10 μg/ml) or pre-immuno IgG (10 μg/ml). A biotinylated goat anti-rabbit IgG secondary antibody was subsequently applied and immunoreactivity was visualized based on streptavidin-biotin-peroxidase with 3,3′-diaminobenzidine as substrate. Hematoxylin counterstain was used to show cellular nucleus. The images were captured using the Q-Imaging digital Camera and Carl Zeiss Axiophot 2 Microscope Integrated Biological Imaging System.
Cell starvation
For starvation experiments, cells were counted before seeding in the plate ensuring same cell number between the scrambled-shRNA cells and TM9SF4-shRNA cell groups. The starvation was carried out in EBSS. If needed, bafilomycin A1 (10 nM) was added in the cell medium for 2 h before EBSS treatment.
LC3 puncta quantification
Cells were transiently transfected with GFP-LC3, or co-transfected with pEGFP-N1 and RFP-LC3, or co-transfected with TM9SF4-GFP and RFP-LC3. Twenty-four hours after the transfection, the cells were starved in EBSS for 2 h with or without bafilomycin A1 (10 nM). Fluorescent images were acquired under Laser Scanning Confocal Imaging System (Fluoview 1000; Olympus, Tokyo, Japan). GFP-LC3 puncta or RFP-LC3 puncta per cell were analyzed using ImageJ software.
Similar measurement was used in RFP-GFP-LC3 puncta quantification. Briefly, the cells carrying scrambled-shRNA or TM9SF4-shRNA were further transfected with RFP-GFP-LC3. Twenty-four hours after transfection, the cells were starved in EBSS for 2 h. GFP/RFP-LC3 puncta per cell were quantified by ImageJ software. The number of yellow puncta (both GFP and RFP) represented autophagosome while red puncta (RFP only) represented autolysosomes.
MTT assay
MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) was used to measure cell metabolic activity. Briefly, the cells with scrambled-shRNA or TM9SF4-shRNA were seeded on the 96-well plate at a density of 4000/well overnight. The cells were then incubated in EBSS for different time duration (2, 4 and 6 h). The cells were then incubated with 250 mg MTT in phenol red free media for 2 h, followed by cell lysis in 10% (w/v) sodium dodecyl sulfate. Purple formazan was dissolved by absolute isopropanol. The absorbance was measured at OD570. The data were expressed as the percent of metabolic activity relative to non-starved cells.
Flow cytometric analysis of cell apoptosis
Apoptosis was determined by flow cytometric analysis of Apopxin Green indicator and 7-amino-actinomycin D (7-AAD) double staining technique by an Apoptosis/Necrosis Detection Kit (Abcam, ab176749) according to the manufacturer’s instructions. 7-AAD labels the nucleus of damaged cells, which can be detected by its red fluorescence (Ex/Em=546/647 nm). Apopxin Green indicator labels phosphatidylserine (PS) on the cell surface of apoptotic cells, which can be measured by green fluorescence (Ex/Em=488/525 nm). Apoptotic cells were expressed as the percentage of early stage (Apopxin Green (+)/7-AAD (−)) plus late stage (Apopxin Green (+)/7-AAD (+)) apoptotic populations. Briefly, the cells were stained with Apopxin Green indicator and 7-AAD, and were then incubated for 30–60 min at room temperature in dark. The level of cell death was examined using a flow cytometer (BD Aria Fusion Cell sorter and Cell Analyzer; BD Sciences, San Jose, CA, USA) and quantified by measuring absorbance at excitation and emission wavelengths of 488 and 525 nm for detection of Apopxin Green Indicator or at excitation and emission wavelengths of 546 and 650 nm for detection of 7-AAD. Quantitative analysis of percentage of apoptotic cells was performed in triplicates.
TUNEL assay
TUNEL staining in tissue sections was conducted using the In Situ Cell Death Detection Kit (Roche, Basel, Switzerland, 11684795910). Briefly, tissue sections were permeabilized in 0.1% Triton X-100 in 0.1% sodium citrate, followed by incubation with the provided fluorescein-conjugated TUNEL reaction mixture in a humidified chamber at 37 °C for 1 h in the dark. The cell nuclei were further stained with 1 μg/ml 4,6 diamidino-2-phenylindole (DAPI) for 30 min at dark in room temperature. The fluorescence was observed under a Olympus FV1000 Confocal Microscope. TUNEL- and DAPI-stained nuclei were analyzed using ImageJ software.
Proximity ligation assay
Protein interactions were detected by Duolink In Situ reagents (Sigma, St. Louis, MO, USA) according to the supplier's protocol. Briefly, the cells were fixed in 4% paraformaldehyde, then permeabilized in 0.1% Triton X-100 for 10 min at room temperature. The slides were incubated with primary antibodies: mouse anti-mTOR (1:50 dilution) and rabbit anti-TM9SF4 (1:50 dilution) at 4 °C overnight. The samples were incubated with secondary antibodies conjugated with PLA probes for 1 h at 37 °C, followed by hybridization and ligation reaction. Rolling-circle amplification was then performed to generate concatemeric fluorescent products, which were visualized as distinct fluorescent dots under a Olympus IX83 inverted microscope. Fluorescent dots per cell were quantified using ImageJ (US National Institutes of Health, Rockville, MD, USA).
Mitochondria membrane potential measurement
Cells were seeded in 96-well plates at a density of 4000 cells/well. On the next day, cells were starved for 2–4 h, and then stained with 50 nM Mitotracker Red FM for 30 min in dark. The excitation wavelength is 581 nm, and the emission peak value at 644 nm was read from a fluorescence SpectraMax i3X Multimode microplate reader (Molecular Devices, Sunnyvale, CA, USA).
Three-dimensional image reconstruction
The three-dimensional (3D) reconstruction was based on the immunostaining images. Z-stack imaging was performed with a × 60 objective using two corresponding lasers (488 and 543 nm). Briefly, the procedure was to set up deep size between 5 and 10 μm dependent on the thickness of samples with a step size of 0.3 μm. Consecutive cross-section images (XYZ) were acquired from the top to the bottom. 3D image reconstruction and display from top view and side view were built up by 3D Olympus Fluoview3.0 software.
TM9SF4 −/− mice
Tm9sf4tm1a(KOMP)Wtsi mice were purchased from KOMP Repository-The Jackson Laboratory (Davis, CA, USA). Tm9sf4tm1a(KOMP)Wtsi were created on a C57BL/6N background and were heterozygous for TM9SF4-knockout. TM9SF4−/− mice were generated by crossing of heterozygous mice. Standard tail DNA genotyping was used to identify TM9SF4−/− mice. The primers for genotyping were: Tm9sf4-forward TTAAAGCCCTAGGAAAGACCAGGGG, Tm9sf4-reverse ACACAGGGGTCTGTGTTATGGTAGC, neoF GGGATCTCATGCTGGAGTTCTTCG. Detailed genotypic information of Tm9sf4tm1a(KOMP)Wtsi can be found at the website: www.komp.org.
Animal starvation
Wild-type or TM9SF4−/− mice were deprived of food but supplied with drinking water for 24 h (14 p.m. to 14 p.m.). Mice were intraperitoneal (i.p.) injected with or without bafilomycin A1 at a dose of 25 ng/g body weight. Mice aged from 8 to 10 weeks were used in this study. All experiments involving animals were performed with the approval by the government of Hong Kong special administrative region department of health under the Animals Ordinance. We followed Guide for Animal Care and Use of Laboratory Animals published by the US National Institute of Health.
Detection of intracellular ROS
The cell permeable fluorescent dye DHE (Beyotime Biotechnology, s0063) was used to determine ROS level in cells and tissues. Briefly, cultured cells were incubated with 5 μM DHE for 30 min at 37 °C in dark, then washed with PBS. DHE fluorescent images were taken by a FV1000 confocal microscope with 488 nm of excitation wavelength and analyzed using ImageJ. For tissue staining, frozen sections of renal tissues were prepared from wild-type or TM9SF4−/− mice. The tissue slides were then subjected to DHE staining, washed with PBS and mounted onto glass slides. The DHE fluorescence analysis is similar to the cultured cells.
Statistical analysis
All data were expressed as mean±S.E.M. Data were statistically analyzed using GraphPad Prism (La Jolla, CA, USA). For experiments with two groups, nonparametric Mann–Whitney U-test was performed. Two-way ANOVA followed by Bonferroni post-test was used for comparison of multiple groups. *P<0.05 was considered to be significant.35, 36
References
Chluba-de Tapia J, de Tapia M, Jaggin V, Eberle AN . Cloning of a human multispanning membrane protein cDNA: evidence for a new protein family. Gene 1997; 197: 195–204.
Fais S, Fauvarque MO . TM9 and cannibalism: how to learn more about cancer by studying amoebae and invertebrates. Trends Mol Med 2012; 18: 4–5.
Froquet R, Cherix N, Birke R, Benghezal M, Cameroni E, Letourneur F et al. Control of cellular physiology by TM9 proteins in yeast and Dictyostelium. J Biol Chem 2008; 283: 6764–6772.
Gao C, Cai Y, Wang Y, Kang BH, Aniento F, Robinson DG et al. Retention mechanisms for ER and Golgi membrane proteins. Trends Plant Sci 2014; 19: 508–515.
Woo CH, Gao C, Yu P, Tu L, Meng Z, Banfield DK et al. Conserved function of the lysine-based KXD/E motif in Golgi retention for endomembrane proteins among different organisms. Mol Biol Cell 2015; 26: 4280–4293.
Benghezal M, Cornillon S, Gebbie L, Alibaud L, Bruckert F, Letourneur F et al. Synergistic control of cellular adhesion by transmembrane 9 proteins. Mol Biol Cell 2003; 14: 2890–2899.
Benghezal M, Fauvarque MO, Tournebize R, Froquet R, Marchetti A, Bergeret E et al. Specific host genes required for the killing of Klebsiella bacteria by phagocytes. Cell Microbiol 2006; 8: 139–148.
Bergeret E, Perrin J, Williams M, Grunwald D, Engel E, Thevenon D et al. TM9SF4 is required for Drosophila cellular immunity via cell adhesion and phagocytosis. J Cell Sci 2008; 121: 3325–3334.
Cornillon S, Pech E, Benghezal M, Ravanel K, Gaynor E, Letourneur F et al. Phg1p is a nine-transmembrane protein superfamily member involved in dictyostelium adhesion and phagocytosis. J Biol Chem 2000; 275: 34287–34292.
Cosson P, Soldati T . Eat, kill or die: when amoeba meets bacteria. Curr Opin Microbiol 2008; 11: 271–276.
Paolillo R, Spinello I, Quaranta MT, Pasquini L, Pelosi E, Lo Coco F et al. Human TM9SF4 is a new gene down-regulated by hypoxia and involved in cell adhesion of leukemic cells. PLoS ONE 2015; 10: e0126968.
Lozupone F, Borghi M, Marzoli F, Azzarito T, Matarrese P, Iessi E et al. TM9SF4 is a novel V-ATPase-interacting protein that modulates tumor pH alterations associated with drug resistance and invasiveness of colon cancer cells. Oncogene 2015; 34: 5163–5174.
Lozupone F, Perdicchio M, Brambilla D, Borghi M, Meschini S, Barca S et al. The human homologue of Dictyostelium discoideum phg1A is expressed by human metastatic melanoma cells. EMBO Rep 2009; 10: 1348–1354.
Lugini L, Matarrese P, Tinari A, Lozupone F, Federici C, Iessi E et al. Cannibalism of live lymphocytes by human metastatic but not primary melanoma cells. Cancer Res 2006; 66: 3629–3638.
Perrin J, Le Coadic M, Vernay A, Dias M, Gopaldass N, Ouertatani-Sakouhi H et al. TM9 family proteins control surface targeting of glycine-rich transmembrane domains. J Cell Sci 2015; 128: 2269–2277.
Klionsky DJ, Emr SD . Autophagy as a regulated pathway of cellular degradation. Science 2000; 290: 1717–1721.
Marino ML, Fais S, Djavaheri-Mergny M, Villa A, Meschini S, Lozupone F et al. Proton pump inhibition induces autophagy as a survival mechanism following oxidative stress in human melanoma cells. Cell Death Dis 2010; 1: e87.
Marino ML, Pellegrini P, Di Lernia G, Djavaheri-Mergny M, Brnjic S, Zhang X et al. Autophagy is a protective mechanism for human melanoma cells under acidic stress. J Biol Chem 2012; 287: 30664–30676.
Rotter D, Rothermel BA . Targets, trafficking, and timing of cardiac autophagy. Pharmacol Res 2012; 66: 494–504.
Ge L, Baskaran S, Schekman R, Hurley JH . The protein-vesicle network of autophagy. Curr Opin Cell Biol 2014; 29: 18–24.
Tan VP, Miyamoto S . Nutrient-sensing mTORC1: integration of metabolic and autophagic signals. J Mol Cell Cardiol 2016; 95: 31–41.
Mizushima N, Yoshimori T, Levine B . Methods in mammalian autophagy research. Cell 2010; 140: 313–326.
Kimura S, Noda T, Yoshimori T . Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 2007; 3: 452–460.
Laplante M, Sabatini DM . mTOR signaling at a glance. J Cell Sci 2009; 122: 3589–3594.
Soderberg O, Gullberg M, Jarvius M, Ridderstrale K, Leuchowius KJ, Jarvius J et al. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods 2006; 3: 995–1000.
Li H, Zhu H, Xu CJ, Yuan J . Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 1998; 94: 491–501.
Puleston D . Detection of mitochondrial mass, damage, and reactive oxygen species by flow cytometry. Cold Spring Harb Protoc 2015; 2015: pdb prot086298.
Johnson MA, Vidoni S, Durigon R, Pearce SF, Rorbach J, He J et al. Amino acid starvation has opposite effects on mitochondrial and cytosolic protein synthesis. PLoS ONE 2014; 9: e93597.
Klionsky DJ, Elazar Z, Seglen PO, Rubinsztein DC . Does bafilomycin A1 block the fusion of autophagosomes with lysosomes? Autophagy 2008; 4: 849–850.
Kimura T, Takabatake Y, Takahashi A, Kaimori JY, Matsui I, Namba T et al. Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J Am Soc Nephrol 2011; 22: 902–913.
Jiang M, Liu K, Luo J, Dong Z . Autophagy is a renoprotective mechanism during in vitro hypoxia and in vivo ischemia-reperfusion injury. Am J Pathol 2010; 176: 1181–1192.
Periyasamy-Thandavan S, Jiang M, Wei Q, Smith R, Yin XM, Dong Z . Autophagy is cytoprotective during cisplatin injury of renal proximal tubular cells. Kidney Int 2008; 74: 631–640.
Lee J, Giordano S, Zhang J . Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling. Biochem J 2012; 441: 523–540.
Lozupone F, Fais S . Cancer cell cannibalism: a primeval option to survive. Curr Mol Med 2015; 15: 836–841.
Skarnes WC, Rosen B, West AP, Koutsourakis M, Bushell W, Iyer V et al. A conditional knockout resource for the genome-wide study of mouse gene function. Nature 2011; 474: 337–342.
Bradley A, Anastassiadis K, Ayadi A, Battey JF, Bell C, Birling MC et al. The mammalian gene function resource: the international knockout mouse consortium. Mamm Genome 2012; 23: 580–586.
Acknowledgements
We thank the Wellcome Trust Sanger Institute Mouse Genetics Project (Sanger MGP) and its funders for providing the mutant mouse line (Tm9sf4), and the distribution center from which we received the mouse line, the KOMP Repository at the University of California, Davis (www.komp.org). Funding and associated primary phenotypic information may be found at www.sanger.ac.uk/mouseportal. We thank Dr. Shi TP for providing the plasmid pCMV6-AV-RFP-LC3 and Dr. Yue JB for providing the plasmid RFP-GFP-LC3. We also thank Dr. Jiang X, Dr. Cao CH, Dr. Chan FL, Dr. Chan AM and Dr. Susan Yung for a variety of human cell lines. This work was supported by grants from the Hong Kong Research Grant Committee CUHK14118516, AoE/M-05/12, RGC-NSFC Joint Grant N_CUHK439/13 and China National Science Foundation Grant 31470912.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by S Fais
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article

Cite this article
Sun, L., Meng, Z., Zhu, Y. et al. TM9SF4 is a novel factor promoting autophagic flux under amino acid starvation. Cell Death Differ 25, 368–379 (2018). https://doi.org/10.1038/cdd.2017.166
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2017.166
This article is cited by
-
Mechanisms and significance of entosis for tumour growth and progression
Cell Death Discovery (2024)
-
CircCEMIP promotes anoikis-resistance by enhancing protective autophagy in prostate cancer cells
Journal of Experimental & Clinical Cancer Research (2022)
-
TM9SF4 is an F-actin disassembly factor that promotes tumor progression and metastasis
Nature Communications (2022)
-
TM9SF4 is a novel regulator in lineage commitment of bone marrow mesenchymal stem cells to either osteoblasts or adipocytes
Stem Cell Research & Therapy (2021)
-
TRPM2 promotes autophagic degradation in vascular smooth muscle cells
Scientific Reports (2020)
