
Abstract
E3 ubiquitin ligases target a growing number of pro- and anti-apoptotic proteins, including tumour suppressor p53, caspases, and the Bcl-2 family. The core apoptosis pathway is well conserved between mammals and Caenorhabditis elegans, but the extent to which ubiquitin ligases regulate apoptotic cell death is not known. To investigate the role of E3 ligases in apoptosis, we inhibited 108 of the 165 predicted E3 ubiquitin ligase genes by RNA interference and quantified apoptosis in the C. elegans germline after genotoxic stress. From this screen, we identified the homologous to E6-associated protein C terminus-domain E3 ligase EEL-1 as a positive regulator of apoptosis. Intriguingly, the human homologue of EEL-1, Huwe1/ARF-BP1/Mule/HectH9, has been reported to possess both pro- and anti-apoptotic functions through its ability to stimulate Mcl-1 and p53 degradation, respectively. Here, we demonstrate that eel-1 is required to promote DNA damage-induced germ cell apoptosis, but does not have a role in physiological germ cell apoptosis or developmental apoptosis in somatic tissue. Furthermore, eel-1 acts in parallel to the p53-like gene cep-1 and intersects the core apoptosis pathway upstream of the Bcl-2/Mcl-1 orthologue ced-9. Although ee1-1 mutants exhibit hypersensitivity to genotoxic stress they do not appear to be defective in DNA repair, suggesting a distinct role for EEL-1 in promoting damage-induced apoptosis in the germline.
Similar content being viewed by others


Main
E3 ubiquitin ligases act at the terminal step of an enzyme cascade that covalently attaches ubiquitin molecules to specific protein substrates. The process of ubiquitination involves either the attachment of a single ubiquitin moiety (monoubiquitination) or several ubiquitins linked in a chain (polyubiquitination) to a target protein. The E3 ligases comprise a large group of proteins subdivided by their domain architecture into the HECT (homologous to E6-associated protein C terminus), RING (really interesting new gene), and U-box (a modified RING motif) families.1 Ubiquitin-based modifications generated by members of these E3 ligase families regulate myriads of biological processes, including cell-cycle progression, differentiation, DNA repair, and programmed cell death (PCD or apoptosis).1 Deregulated expression of E3 ligases can result in a number of pathological conditions such as neurodegeneration and cancer. For example, endogenous levels of tumour suppressor p53 are under strict regulation by the RING-finger E3 ubiquitin ligase Mdm2 (murine double minute clone 2 oncoprotein), which targets p53 for ubiquitin-mediated proteolysis under normal conditions.2 In response to stresses such as DNA damage, p53 and Mdm2 become phosphorylated and their interaction is inhibited, which leads to stabilization and activation of p53. In addition to Mdm2, other classes of E3 ligases including Huwe1, COP1, and Pirh2 have all been shown to control the stability of p53 in cells, underscoring the importance of carefully regulating the levels of this tumour suppressor.2 Beyond p53, E3 ubiquitin ligases regulate a number of anti- and pro-apoptotic proteins such as the inhibitor of apoptosis proteins, the Bcl-2 family, and caspases.3, 4, 5, 6, 7 Given the emerging roles for ubiquitination in development and disease there remains much to be learned about the extent to which E3 ubiquitin ligases regulate apoptosis.
The nematode Caenorhabditis elegans is an excellent model organism for studying the role of ubiquitination in apoptosis. This organism has a well-conserved apoptosis signalling pathway that is required for almost all PCDs and a highly conserved complement of E3 ubiquitin ligases.8, 9 C. elegans contains a single p53-like gene, cep-1, which is required to activate apoptosis of germ cells treated with DNA-damaging agents.10, 11 Like mammals, C. elegans sense damaged DNA through a conserved checkpoint that includes the 9-1-1 complex (hpr-9, hus-1, and mrt-2) and the Tel2 orthologue clk-2. These genes are required to initiate a transient cell-cycle arrest of mitotically proliferating germ cells and/or activate cep-1-dependent apoptosis in the pachytene region of the germline.12, 13 We previously discovered a role for the Skp1/cullin/F-box (SCF) E3 ubiquitin ligase SCFFSN-1 in the negative regulation of cep-1-dependent germ cell apoptosis.14 Interestingly, the human equivalent of SCFFSN-1 (SCFFBXO45) was subsequently shown to have a similar role in mammalian cell apoptosis by promoting the proteasome-dependent degradation of the p53 paralogue p73, highlighting the functional conservation of these ligases.15 The simplified structure of the C. elegans cell death pathway and the ease in which genes can be inhibited by RNA interference (RNAi) make this organism an ideal system to study the regulation of DNA damage-induced apoptosis.
To comprehensively examine the role of C. elegans ‘ubiquitome’ in damage-induced apoptosis, we systematically inhibited 108 of the 165 predicted RING, HECT, and U-box-containing E3 ligase genes by RNAi and quantified ionizing radiation (IR)-induced apoptosis in the germline. From this screen, we identified the HECT-domain E3 ligase EEL-1, a homologue of human Huwe1/ARF-BP1/Mule, as a positive regulator of IR-induced germline apoptosis. We show that eel-1 regulates IR-induced germline apoptosis, but is dispensable for physiological germ cell apoptosis and developmental apoptosis in the soma. EEL-1 promotes apoptosis by a mechanism that does not affect the transcriptional activity of the p53-like protein CEP-1 or alter endogenous levels of the Mcl-1-like protein CED-9. Although eel-1 mutants exhibit a normal checkpoint response, their progeny are hypersensitive to IR and replication inhibitors. Unexpectedly, the sensitivity of eel-1 embryos to radiation is not due to an overt defect in DNA repair based on the accumulation of RAD-51 foci in parental germ cells. We conclude that EEL-1 fine-tunes DNA damage-induced apoptosis signalling in the C. elegans germline.
Results
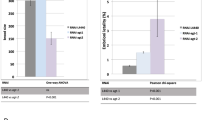
The C. elegans genome contains a total of 165 predicted E3 ubiquitin ligases, encoding 152 RING proteins, 9 HECT proteins, and 4 U-box proteins.9 To determine the extent to which these E3 ligases regulate DNA damage-induced germline apoptosis, we systematically inhibited 108 of these 165 genes by RNAi and quantified germline apoptosis after exposure to 60 Gy of IR. From this screen, we identified wwp-1 and eel-1, two genes encoding HECT-domain containing E3 ligases that caused resistance to IR-induced germline apoptosis when inhibited by RNAi (Figures 1a and b). The wwp-1 gene has been shown to have a role in nucleotide excision repair in C. elegans, and the human homologue WWP1 has been reported to negatively regulate both p53 and p63.16, 17, 18 However, as we were unable to recapitulate the germline apoptosis phenotype using three mutant alleles of wwp-1 (gk372, gk397, and ok1102, data not shown) we did not follow up on this gene and instead focused on eel-1.

eel-1 and wwp-1 are required for promoting IR-induced germ cell apoptosis. (a) Wild-type animals fed eel-1(RNAi) (white bars) are resistant to germline apoptosis after treatment with 60 Gy of IR compared with animals fed control RNAi (black bars). (b) wwp-1(RNAi) (white bars) also caused resistance to germ cell apoptosis in response to 60 Gy of IR compared with controls. Animals were exposed to IR at the young adult stage and germ cell apoptosis was quantified 24 h later. (c and d) Synchronized L4 hermaphrodites of the indicated genotype were subjected to increasing doses of IR and germ cell corpses were quantified 24 h later. Data represent mean±S.E.M. of at least three independent experiments. Between 15 and 30 germlines were scored per experiment
We confirmed our initial eel-1(RNAi) observation with two different deletion mutants, which exhibited similar levels of resistance to IR-induced germline apoptosis over a range of dosages (Figures 1c and d). These results suggest that eel-1 acts a positive regulator of damage-induced apoptosis in the C. elegans germline. In the absence of DNA damage, ∼50% of germ cells undergo physiological apoptosis.19 We noticed that physiological germ cell apoptosis was similar in eel-1 mutants compared with wild-type controls (Figures 1c and d), suggesting that eel-1 specifically regulates damage-induced germ cell death. There are two possible explanations for how eel-1 regulates damage-induced germ cell death. The first is that eel-1 may control the timing of apoptosis such that germ cell corpses do not appear in eel-1 mutants until after the 24 h time point used for our analysis. Alternatively, eel-1 may be required to activate damage-induced germ cell apoptosis similar to the p53-like gene cep-1.10, 11 To test the first possibility, we performed a time course analysis and found that eel-1(ok1575) mutants were resistant to IR-induced apoptosis, even 48 h after treatment (Figure 2a). This suggests that eel-1 is required to promote damage-induced germ cell apoptosis rather than control the kinetics of corpse appearance. As lower levels of IR-induced germ cell apoptosis could be the result of decreased germ cell proliferation in eel-1 mutants, we determined the number of nuclei in the pachytene region of the germline. The eel-1 mutants had similar numbers of nuclei in their pachytene regions as wild-type controls (Supplementary Table 1), suggesting that lower levels of apoptosis were not due to a proliferation defect. This is supported by our observation that there are similar numbers of mitotically proliferating germ cells in eel-1 mutants and wild-type animals (Figures 3a and b). To verify that eel-1 mutants do not have a general defect in apoptosis, we quantified corpses using the engulfment defective ced-1(e1735) allele.20 In ced-1(e1735); eel-1(ok1575) double mutants the levels of physiological germline apoptosis were similar to ced-1(e1735) single mutants over the same time course (Figure 2b). To determine if eel-1 regulates developmental apoptosis in the soma, we compared corpse numbers in developing ced-1(e1735) embryos with ced-1(e1735); eel-1(ok1575) embryos. Similar to physiological germline apoptosis, we found no difference in the number of persistent corpses in the embryos and first stage larvae of ced-1; eel-1 versus ced-1 mutants (Figure 2c). Collectively, these results indicate that eel-1 specifically regulates DNA damage-induced germ cell apoptosis.

eel-1 specifically promotes DNA damage-induced germ cell apoptosis. (a) L4 stage hermaphrodites of the indicated genotype were subjected to either 0 or 60 Gy of IR and germ cell corpses were quantified at the indicated times after irradiation. Data represent mean±S.E.M. of at least three independent experiments. (b) Physiological germ cell apoptosis is unaffected by mutation of eel-1. Germ cell corpses were quantified in strains carrying the ced-1(e1735) engulfment defective mutation. Between 15 and 30 germlines were scored per experiment in panels (a) and (b). (c) eel-1 does not regulate developmental apoptosis. Corpses were quantified at specific stages of embryonic development and first stage (L1) larvae. Around seven animals were scored per stage. Data represent mean±S.E.M.

eel-1 mutants are not defective in DNA damage checkpoint function. (a) eel-1(ok1575) mutants undergo a transient arrest in proliferation of mitotic germ cells in response to IR that is indistinguishable from wild-type or cep-1(gk138) mutants. Germlines are outlined in solid black lines and arrows indicate normal mitotic nuclei. Dotted arrows indicate enlarged mitotic nuclei 20 h after exposure to 120 Gy of IR. (b) Quantification of the number of nuclei per unit area in the mitotic region of the germline as shown in panel a. Data represent mean±S.E.M. of two independent experiments. Between 10 and 15 germlines were scored per experiment. (c) Sensitivity of L1 stage eel-1 mutants to IR-induced sterility is similar to wild type. Twenty animals of the indicated genotype were irradiated at the L1 stage and fertility assessed after they developed into adults
In wild-type animals genotoxic stress activates a conserved DNA damage checkpoint, which causes a transient arrest in mitotically proliferating germ cells and activation of DNA repair. The checkpoint genes hpr-9/RAD9, hus-1/HUS1, mrt-2/RAD1, and clk-2/TEL2 are required for cell-cycle arrest and activation of DNA repair.12, 13 Loss-of-function mutations in these genes also cause resistance to germline apoptosis because the checkpoint is unable to activate the p53-like protein CEP-1. As reduced levels of apoptosis in eel-1 mutants could be the result of defective checkpoint activation, we next examined the checkpoint response in wild-type, cep-1(gk138), and eel-1(ok1575) animals. Contrary to van Haaften et al.21 who reported a mild cell-cycle arrest defect when eel-1 was knocked down by RNAi, we observed a robust arrest of mitotic germ cell proliferation in eel-1 deletion mutants that was indistinguishable from wild-type controls (Figures 3a and b). In addition to defective cell-cycle arrest, checkpoint mutants also generate higher frequencies of inviable progeny after exposure to genotoxic agents because they cannot properly engage DNA repair pathways.12, 13 Since Huwe1 has recently been shown to regulate DNA repair in mammalian cells,22 we determined if eel-1 mutants were sensitive to DNA damage by two methods. First, we quantified inviable progeny generated by adults exposed to DNA-damaging agents; second, we determined the frequency of animals irradiated at the L1 stage that developed into sterile adults.23 We found that eel-1 mutants generated more inviable progeny than wild-type controls after treatment with IR (Table 1) or replication inhibitors (Supplementary Table 2). In contrast, the percentage of irradiated L1 animals that became sterile by the adult stage was the same as wild type, whereas clk-2 mutants were hypersensitive (Figure 3c). This apparently contradictory result could be explained if eel-1 germlines are not hypersensitive to DNA-damaging agents during larval development but become sensitive by the adult stage.
To address the potential repair function of EEL-1 in more detail, we dissected germlines of irradiated eel-1 adults and stained with antibodies to the double-strand break repair protein RAD-51. RAD-51 forms foci at sites of double-strand breaks in the germline that increase after treatment with IR and persist for longer periods of time in the germlines of adult checkpoint mutants.24, 25 We found that the number of persistent RAD-51 foci in eel-1 mutants was similar to wild-type controls before and after treatment with IR (Figure 4). Interestingly, we observed more RAD-51 foci in irradiated cep-1; eel-1 double mutants (Figure 4h), suggesting that the combined ablation of these two genes causes defects in DNA repair. However, we cannot exclude the possibility that this could be due to a general metabolic defect in cep-1; eel-1 mutants as they also generate more inviable progeny in the absence of genotoxic stress (data not shown). Taken together, the above results suggest that eel-1 likely acts downstream or independently of the DNA damage checkpoint.

No persistent RAD-51 foci in eel-1 mutants. RAD-51 foci were scored 24 h after exposure to 30 Gy of IR according to Alpi et al.24 Representative images of RAD-51 foci (a–h) and DAPI-stained nuclei (a′–h′) are shown for 0 Gy (a–d and a′–d′) and after 30 Gy (e–h and e′–h′) for wild-type (a, a′, e and e′), eel-1(ok1525) mutants (b, b′, f and f′), cep-1(gk138) mutants (c, c′, g and g′), and cep-1(gk138); eel-1(ok1525) double mutants (d, d′, h and h′)
As cep-1/p53 is required to activate damage-induced germline apoptosis and Huwe1 has been shown to negatively regulate p53 in mammalian cells, we next asked whether the resistance to IR-induced apoptosis in eel-1 mutants was the result of decreased CEP-1 transcriptional activity. We previously showed that the E3 ubiquitin ligase SCFFSN-1 negatively regulates IR-induced apoptosis through CEP-1, a finding which was subsequently shown to be conserved in humans.14, 15 To determine if EEL-1 regulates CEP-1 activity, we quantified the transcript abundance of its pro-apoptotic target gene egl-1 by RT-PCR. Following irradiation, levels of egl-1 transcript increased by approximately nine-fold in wild-type animals and eel-1(ok1575) mutants (Figure 5), suggesting that eel-1 is not required for CEP-1 activation and likely modulates damage-induced germline apoptosis downstream or independently of cep-1.

eel-1 does not affect the transcriptional activity of the p53-like protein CEP-1. The abundance of the CEP-1 target gene egl-1 was similar in eel-1(ok1575) mutants as wild-type controls, but not induced by IR in cep-1(gk138) mutants. Data represent mean±S.E.M. of two independent experiments
To determine where eel-1 acts in relation to the core apoptosis signalling pathway, we examined the epistatic relationship between eel-1 and the pro-survival gene ced-9 by constructing a double-mutant strain between the ced-9(n2812) putative null allele26 and eel-1(ok1575). As CED-9 functions to protect cells from undergoing apoptosis, loss of CED-9 causes massive germ cell death in adult hermaphrodites. In the absence and presence of damage, homozygous ced-9(n2812) hermaphrodites generated from heterozygous mothers had significantly increased levels of germ cell apoptosis compared with wild type, which was not suppressed by the loss of eel-1 (Figure 6a). This is consistent with a model where eel-1 acts upstream or independently of ced-9. Complete ablation of ced-9 in homozygous mutants resulted in similar levels of germline apoptosis in the absence versus presence of IR (Figure 6a). Therefore, if the role of EEL-1 is to specifically regulate CED-9 in response to DNA damage this effect might be masked when the ced-9 gene is completely ablated. To address this possibility we took two approaches: first, IR-induced germline apoptosis was quantified in a strain heterozygous for the ced-9(n2812) allele but homozygous for eel-1(ok1575); second, the levels of endogenous CED-9 protein were examined in eel-1 mutants by western blotting. Reduction of ced-9 by one copy in ced-9(n2812)/hT2 animals resulted in a modest increase in physiological germline apoptosis and hypersensitivity to IR (Figure 6b). In ced-9(n2812)/hT2; eel-1(ok1575) animals, IR increased germline apoptosis to levels that were similar to those observed in ced-9(n2812)/hT2 heterozygotes, suggesting that eel-1 acts upstream or independently of ced-9 (Figure 6b). In light of published results that showed Huwe1 to target Mcl-1 (a CED-9 homologue) for degradation in response to DNA damage,7 we also quantified the level of CED-9 protein in wild-type animals and eel-1 mutants before and after DNA damage. There was no significant difference in CED-9 protein levels in eel-1(ok1575) or eel-1(zu462) mutants compared with wild-type controls following treatment with IR (Figure 6c), supporting a model where EEL-1 does not target CED-9 for degradation in C. elegans. Interestingly, we found CED-9 protein levels to be unchanged in unirradiated wild-type animals compared with irradiated worms, suggesting that CED-9 stability does not change in response to DNA damage. However, it is possible that CED-9 protein is regulated by DNA damage only in the pachytene region of the germline, which might not be detectable by western analysis from whole-worm lysates.

eel-1 acts upstream or independently of ced-9. (a and b) Hermaphrodites of the indicated genotypes were irradiated with 60 Gy of IR at the L4 larval stage and germ cell apoptosis quantified 24 h later. Data represent mean±S.E.M. of three independent experiments. Between 15 and 30 germlines were scored per experiment. (c) Endogenous CED-9 protein levels are not affected by eel-1 status or IR. Hermaphrodites of the indicated genotype were treated with 120 Gy of IR at the L4 stage and the protein was extracted from whole-worm lysates after 24 h. Western analysis was performed using an anti-CED-9 antibody kindly provided by Dr. Barbara Conradt. The protein α tubulin was used as a loading control. Quantification was performed using ImageJ software. Protein abundance is relative to wild type at the indicated dose of IR
As eel-1 appears to function downstream of cep-1 and does not regulate CED-9 protein levels, we sought to identify other possible substrates of EEL-1. Previous work revealed a role for EEL-1 in cell fate specification via the transcription factor SKN-1, which regulates mesendodermal cell fate specification in the early embryo and is the only proposed substrate of EEL-1 in C. elegans.27 As EEL-1 and SKN-1 have been proposed to interact in the early embryo to regulate development, it is possible that they may also act in the same pathway to regulate apoptosis in the germline. If SKN-1 is a target of EEL-1 in germ cell apoptosis signalling, ablation of skn-1 might increase sensitivity to DNA damage-induced germ cell apoptosis. While ablation of skn-1 by RNAi and a loss-of-function mutation in skn-1 caused the expected embryonic lethality in the next generation, IR-induced germ cell apoptosis was indistinguishable from wild-type controls (Supplementary Figure 1, and data not shown). These results suggest that skn-1 does not have a major role in DNA damage-induced germ cell apoptosis and that EEL-1 likely targets a novel substrate.
Discussion
We screened 65% of the 165 predicted E3 ligase genes comprising the HECT, RING, and U-box domain subgroups and identified a pro-apoptotic role for eel-1. We also uncovered the WWP1 homologue (wwp-1) in this screen and found a similar pro-apoptotic role as eel-1 in germline apoptosis. However, as existing deletion alleles of wwp-1 did not recapitulate the apoptotic phenotypes observed by RNAi we focused on characterizing eel-1. The mammalian homologue of EEL-1, Huwe1 (also known as ARF-BP1, HectH9, or Mule) has been shown to regulate both pro-apoptotic (p53) and anti-apoptotic (Mcl-1) proteins, as well as the DNA repair proteins Cdc6 and DNA polymerase β.7, 22, 28, 29 Our results suggest that EEL-1 regulates apoptosis in C. elegans by a different mechanism than its mammalian counterpart. As there is a robust cell-cycle arrest and the pro-apoptotic gene egl-1 is induced to wild-type levels in eel-1 mutants exposed to IR (Figures 3 and 5), our results imply that eel-1 functions in a parallel signalling pathway to promote damage-induced germline apoptosis. Consistent with this, several recent reports have uncovered genes that act independently or downstream of cep-1 to promote apoptosis, such as the Sirtuin sir-2.1, the Rb gene lin-35, the inhibitor of growth homologue ing-3 and the Krit1/Ccm1 gene kri-1.30, 31, 32, 33 Like eel-1, all of these genes have pro-apoptotic function independent of the DNA damage checkpoint or CEP-1 transcriptional activity. In C. elegans, EEL-1 has been shown to bind and possibly stimulate degradation of the transcription factor SKN-1 in the early embryo.27 We did not observe any changes in IR-induced germline apoptosis after knocking down skn-1 by RNAi or in skn-1 mutants (Supplementary Figure 1), suggesting that EEL-1 targets a novel substrate to regulate apoptosis.
The hypersensitivity of eel-1 mutants to IR-induced lethality (Table 1) is intriguing in light of a recent study implicating Huwe1 in the regulation of base excision repair by targeting DNA polymerase β.22. Interestingly, clk-2 checkpoint mutants are also hypersensitive to IR and replication stress and exhibit synthetic lethal interactions with cep-1, suggesting that cep-1 cooperates with clk-2 to modulate replication stress during embryonic development.12, 34 However, there was no difference in the accumulation and persistence of RAD-51 foci in the germlines of eel-1 mutants compared with wild-type controls exposed to IR (Figure 4). We conclude that either EEL-1 has no role in DNA repair or it regulates repair independent of RAD-51 accumulation. Interestingly, cep-1; eel-1 double mutants generate about 40% lethal progeny in the absence of exogenous DNA damage, similar to cep-1; clk-2 double mutants,34 and they appear to have more intense RAD-51 foci after treatment with IR (Figure 4). Thus, it is possible that these genes have a cooperative role in DNA repair. The hypersensitivity of eel-1 mutants to the replication inhibitor hydroxyurea (HU) (Supplementary Table 2) provides additional evidence that this E3 ligase may target a unique substrate downstream of the checkpoint to coordinate repair and apoptosis. However, strains carrying either allele of eel-1 exhibit a range of pleiotropic phenotypes, such as a protruding vulva and egg laying defects. Thus, it is possible that eel-1 mutants are hypersensitive to stresses that simply antagonize an already tenuous existence. As Huwe1 has been shown to target multiple proteins that regulate apoptosis and DNA repair in mammals, it is possible that EEL-1 also targets multiple substrates in C. elegans. Consistent with this, EEL-1 has been implicated in the control of cell fate specification through SKN-1 and more recently in hemidesmosome maturation in C. elegans.27, 35 Therefore, ablation of the eel-1 gene likely deregulates several cellular proteins and signalling pathways that could indirectly cause animals to become hypersensitive to stresses such as DNA damage. More work will have to be done to determine whether EEL-1 has a direct role in DNA repair. However, we believe that eel-1 has a specific role in the regulation of DNA damage-induced apoptosis as there are no proliferation defects, no changes in physiological germline apoptosis, and no effects on developmental apoptosis in eel-1 mutants. As IR-induced apoptosis in eel-1; ced-9 double mutants is the same as ced-9 single mutants (Figure 6) we conclude that EEL-1 does not likely affect the core apoptotic pathway downstream of egl-1. This is supported by the absence of defects in physiological germline apoptosis or developmental apoptosis in eel-1 mutants (Figure 2).
How might eel-1 regulate DNA damage-induced germline apoptosis? Taken together, our data suggest that eel-1 regulates apoptosis downstream or independently of cep-1 and the DNA damage checkpoint (Figure 7). It is unlikely that EEL-1 targets CED-9 directly, as changes in endogenous CED-9 levels (or mobility shifts) were not observed in eel-1 mutants following treatment with IR (Figure 6). However, as CED-9 is expressed in most cells of the organism it is possible that alterations in the pachytene region of the germline might not be detectable by western analysis of whole worm lysates. In general, polyubiquitin chains generated through linkages at residues K11, K24, or K48 cause substrates to be directed for degradation by the proteasome. On the other hand, monoubiquitination or polyubiquitination via linkage at K63 can alter the activity and/or subcellular localization of proteins.1 As the BH3-only gene egl-1 is also dispensable for physiological germ cell apoptosis,19 it is possible that EEL-1 could monoubiquitinate EGL-1 (or an EGL-1 accessory protein) to control its activity and/or subcellular localization in response to DNA damage. It is well established that egl-1 is upregulated transcriptionally to promote apoptosis during development and in the germline in response to genotoxic stress.8, 36, 37 However, it is not known whether EGL-1 is regulated by post-translational modifications in C. elegans. Post-translational modification by ubiquitin has been shown to have a role in the regulation of BH3-only proteins in mammalian cells. For example, the BH3-only protein BIM, which is responsible for the apoptosis of lymphocytes, myeloid cells, and neurons, is ubiquitinated and degraded in human osteoclasts by the c-Cbl family of E3 ubiquitin ligases.38 It will be important to determine whether this aspect of BH3-only protein regulation is conserved in C. elegans, which can be assessed once antibodies to EGL-1 become available. Although further studies are required to elucidate the biochemical mechanism by which EEL-1 regulates damage-induced germline apoptosis, our results suggest that this ligase may have an ancient role in apoptosis signalling that operates by a distinct mechanism in C. elegans.

Model for EEL-1 regulation of IR-induced germ cell apoptosis in C. elegans. EEL-1 may act at the level of EGL-1 or CED-9. Hypothetical relationships are indicated by dashed arrows. The corresponding mammalian pathway and the EEL-1 orthologue Huwe1 are shown on the right
Materials and Methods
General methods and strains
All C. elegans strains used were cultured at 20°C on nematode growth media (NGM) plates according to standard protocols.39 The wild-type strain used was Bristol N2. The following strains were provided by the Caenorhabditis Genetics Centre (CGC): ced-1(e1735)I, cep-1(gk138)I, wwp-1(gk372)I, wwp-1(ok1102)I, wwp-1(gk397)I, dpy-5(e61) unc-13(e51)/hT2[bli-4(e937) let-?(q782) qIs48]I, hus-1(op244) I/hT2[bli-4(e937) let-?(q782) qIs48] (I;III), clk-2(qm37)III, eel-1(ok1575)IV, and eel-1(zu462) unc-33(e204)IV. The strain ced-9(n2812)/qC1 III was kindly provided by Dr. Robert Horvitz. The strain ced-9(n2812)/hT2 III was generated with dpy-5(e61) unc-13(e51)/hT2[bli-4(e937) let-?(q782) qIs48]I and ced-9(n2812)/qC1 III using standard genetic techniques. The double mutants ced-9(n2812)/hT2 III; eel-1(ok1575)IV and ced-1(e1735)I, eel-1(ok1575)IV were generated using standard methods. The linked unc-33(e204) mutation was segregated away from eel-1(zu462) unc-33(e204)IV to generate eel-1(zu462)IV.
Screening and RNAi
RNAi experiments were performed by the feeding method and all of the RNAi bacterial strains were obtained from the Ahringer RNAi library as we previously described.14 A bacterial strain expressing the empty RNAi vector L4440 was used as a control. To screen the 108 E3 ligase genes represented in the Ahringer RNAi library for defects in IR-induced germline apoptosis, wild-type animals of the fourth larval stage (L4) were fed double-stranded RNA expressing bacteria and were allowed to lay eggs for 3–4 days. Young adult animals of the F1 generation were treated with 60 Gy of IR and germline apoptosis was quantified as described (see below). All 108 E3 ligases available in the RNAi library were screened at least once.
C. elegans germline apoptosis
Synchronized hermaphrodites at the L4 stage of development were exposed to various doses of IR (137Cs source) and were allowed to recover on NGM plates at 20°C. Germ cell apoptosis was quantified in the pachytene region of one gonad arm per animal using Nomarski optics 24 h post-irradiation as previously described.10 As older eel-1 mutants exhibit Pvl and Egl phenotypes, we scored germline apoptosis in worms 24 h after the L4molt. Developmental apoptosis in staged embryos and physiological germ cell apoptosis in young adult hermaphrodites were quantified in strains carrying the ced-1(e1735) mutation to prevent corpse engulfment.19
Cell-cycle arrest and radiation sensitivity
To determine the effects of IR on germ cell proliferation, synchronized L4 animals were treated with 120 Gy of IR and cell nuclei per area in the mitotic region of the germline were quantified 24 h after irradiation. Analysis was performed at × 630 using Nomarski microscopy and OpenLab software (PerkinElmer, Waltham, MA, USA), as previously described.40
To evaluate radiation sensitivity, L4-stage hermaphrodites were treated with either 60 or 120 Gy of IR. Approximately 12 h later, irradiated worms and unirradiated controls were transferred to new plates and allowed to lay eggs for 5 h (eggs laid during this time correspond to germ cells irradiated at pachytene). The adults were removed and the number of unhatched eggs were counted 24 h later. Radiation-induced sterility was evaluated by exposing L1 stage hermaphrodites to IR and quantifying the percentage of animals that developed into sterile adults.23
Characterization of eel-1 mutants
To determine if ok1575 and zu462 are recessive alleles, we crossed these eel-1 mutants with wild-type animals and quantified damage-induced germ cell apoptosis in the heterozygous progeny. The eel-1 heterozygous mutants had similar levels of germ cell apoptosis as wild-type controls treated with 60 Gy of IR (data not shown), indicating that ok1575 and zu462 are recessive alleles.
Both eel-1(ok1575) and eel-1(zu462) mutants also display some pleiotropic defects such as protruding vulvas, slow growth, and egg-laying defects. It is unknown whether these are null alleles, as no deficiencies are available that remove the eel-1 locus. The eel-1(ok1575) allele contains a 1.6-kb in-frame deletion and RT-PCR analysis revealed that a truncated eel-1 transcript is expressed in these animals (data not shown). The eel-1(zu462) mutant isolated by Page et al.27 contains a 1.9-kb deletion followed by a 5.9-kb inverted duplication that is predicted to encode an extra 36 amino acids and generate a truncated protein of 3911 amino acids.
egl-1 quantitative real-time PCR
Approximately 2000 synchronized L4 stage animals were treated with either 0 or 120 Gy of IR and RNA was isolated 24 h later. Worms were washed twice in M9 buffer and stored at −80°C overnight in 1 ml of Trizol (Invitrogen Canada, Burlington, ON, Canada). The next day lysates were thawed and RNA was extracted with 0.2 ml of chloroform then centrifuged at 15 000 r.p.m. for 15 min at 4°C. To precipitate RNA, 200 μl of supernatant was placed in a fresh 1.5-ml microcentrifuge tube plus 500 μl of isopropanol then centrifuged at 15 000 r.p.m. for 15 min. The pellet was washed with 1 ml of 75% ethanol in DEPC H2O (Invitrogen) and centrifuged at 15 000 r.p.m. for 5 min. The pellet was air dried for 15 min then re-suspended in 25 μl DEPC H2O.
A cDNA template was generated using 2 μg of RNA, as previously described.39 The egl-1 transcript was amplified using primers described by Hofmann et al.36 Amplification of tubulin transcript was used as a control. Real-time monitoring of amplification was achieved using a 7900HT real-time PCR system (Applied Biosystems, Foster City, CA, USA).
Western blot analysis of CED-9 protein levels
Approximately 2000–3000 synchronized hermaphrodites at the L4 stage of development were exposed to either 0 or 120 Gy of IR. Animals were lysed by sonication in SDS lysis buffer (4% SDS, 100 mM Tris-HCl (pH 6.8), 20% glycerol) 24 h after treatment with IR. In total, 30 μg of soluble protein was suspended in SDS loading buffer (4% SDS, 10% 2-mercaptoethanol, 20% glycerol, 0.004% bromophenol blue, 100 mM Tris-HCl (pH 6.8)) and was boiled for 4 min and then centrifuged at 12 000 × g for 1 min. Proteins were separated by SDS-PAGE, transferred onto a nitrocellulose membrane, and immunoblotted with a 1/1000 dilution of rabbit anti-CED-9 antibody (kindly provided by Dr. Barbara Conradt) or a 1/2000 dilution of mouse anti-α-tubulin antibody (Sigma-Aldrich Canada Ltd, Oakville, ON, Canada). ImageJ software (http://rsbweb.nih.gov/ij/index.html) was used to analyse bands corresponding to CED-9 and α-tubulin and quantification was performed by normalizing CED-9 protein levels to α-tubulin protein levels.
Immunocytochemistry
Worms at the young adult stage were exposed to either 0 or 30 Gy of IR and allowed to recover for 24 h at 20°C. Irradiated or unirradiated control hermaphrodites were washed once in PBS, resuspended in 30 μl of PBS+0.2 mM tetramisole on polylysine-coated slides. Germlines were extruded by removing the heads and tails using scissoring motion with two 27-G needles. Tetramisole was replaced with 1% paraformaldehyde in PBS for 10 min at room temperature followed by freeze cracking on dry ice for 1 min. Post fixation was carried by immersing samples in a 1 : 1 mixture of methanol/acetone at −20°C for 1 min. Samples were then permeabilized with PBS+1% Triton X-100 for 10 min at room temperature, then washed three times with PBS+0.1% Tween 20 (PBST) for 10 min per wash. To reduce background, samples were incubated with Image-iT FX signal enhancer (Invitrogen) for 20 min, then incubated for 30 min in PBS+0.1% Tween 20+1% BSA (PBSTB). RAD-51 antibodies (SDI, cat no. 2948.00.02) were diluted 1/100 in PBSTB and incubated at 4°C overnight in a humid chamber. Samples were washed three times in PBST then samples were incubated in Alexa 488 (Invitrogen) secondary antibody diluted 1/1000 in PBSTB for 1 h at room temperature. After washing four times for 15 min in PBST, the samples were mounted in prolong Gold (Invitrogen). DAPI diluted 1/1000 from 1 mg/ml stock solution was added at the second wash step.
HU sensitivity
NGM plates were prepared with 10 mM HU in the media as described previously.34 Hermaphrodites at the L4 stage were placed on HU supplemented NGM plates and allowed to develop into young adults for 24 h then transferred to fresh HU plates and allowed to lay eggs for 6 h. The adults were then removed and the deposited eggs were counted. Unhatched eggs were counted after 48 h and the total number of living larvae and adult animals were counted after 96 h. Percent lethality was determined in triplicate.
Abbreviations
- HECT:
-
homologous to E6-associated protein C terminus
- RING:
-
really interesting new gene
- PCD:
-
programmed cell death
- Mdm2:
-
murine double minute clone 2 oncoprotein
- IAP:
-
inhibitor of apoptosis protein
- SCF:
-
Skp-Cullin-F-box
- IR:
-
ionizing radiation
- NGM:
-
nematode growth media
- RNAi:
-
RNA interference
- HU:
-
hydroxyurea
- IPTG:
-
isopropyl-β-D-thiogalactopyranoside
References
Pickart CM . Mechanisms underlying ubiquitination. Annu Rev Biochem 2001; 70: 503–533.
Brooks CL, Gu W . p53 ubiquitination: Mdm2 and beyond. Mol Cell 2006; 21: 307–315.
Dimmeler S, Breitschopf K, Haendeler J, Zeiher AM . Dephosphorylation targets Bcl-2 for ubiquitin-dependent degradation: a link between the apoptosome and the proteasome pathway. J Exp Med 1999; 189: 1815–1822.
Li B, Dou QP . Bax degradation by the ubiquitin/proteasome-dependent pathway: involvement in tumor survival and progression. Proc Natl Acad Sci USA 2000; 97: 3850–3855.
Marshansky V, Wang X, Bertrand R, Luo H, Duguid W, Chinnadurai G et al. Proteasomes modulate balance among proapoptotic and antiapoptotic Bcl-2 family members and compromise functioning of the electron transport chain in leukemic cells. J Immunol 2001; 166: 3130–3142.
Yang Y, Fang S, Jensen JP, Weissman AM, Ashwell JD . Ubiquitin protein ligase activity of IAPs and their degradation in proteasomes in response to apoptotic stimuli. Science 2000; 288: 874–877.
Zhong Q, Gao W, Du F, Wang X . Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell 2005; 121: 1085–1095.
Conradt B, Xue D . Programmed cell death. WormBook 2005; 6: 1–13.
Kipreos ET . Ubiquitin-mediated pathways in C. elegans. WormBook 2005: 1–24.
Derry WB, Putzke AP, Rothman JH . Caenorhabditis elegans p53: role in apoptosis, meiosis, and stress resistance. Science 2001; 294: 591–595.
Schumacher B, Hofmann K, Boulton S, Gartner A . The C. elegans homolog of the p53 tumor suppressor is required for DNA damage-induced apoptosis. Curr Biol 2001; 11: 1722–1727.
Ahmed S, Alpi A, Hengartner MO, Gartner A . C. elegans RAD-5/CLK-2 defines a new DNA damage checkpoint protein. Curr Biol 2001; 11: 1934–1944.
Gartner A, Milstein S, Ahmed S, Hodgkin J, Hengartner MO . A conserved checkpoint pathway mediates DNA damage-induced apoptosis and cell cycle arrest in C. elegans. Mol Cell 2000; 5: 435–443.
Gao MX, Liao EH, Yu B, Wang Y, Zhen M, Derry WB . The SCFFSN-1 ubiquitin ligase controls germline apoptosis through CEP-1/p53 in C. elegans. Cell Death Differ 2008; 15: 1054–1062.
Peschiaroli A, Scialpi F, Bernassola F, Pagano M, Melino G . The F-box protein FBXO45 promotes the proteasome-dependent degradation of p73. Oncogene 2009; 28: 3157–3166.
Astin JW, O’Neil NJ, Kuwabara PE . Nucleotide excision repair and the degradation of RNA pol II by the Caenorhabditis elegans XPA and Rsp5 orthologues, RAD-3 and WWP-1. DNA Repair (Amst) 2008; 7: 267–280.
Laine A, Ronai Z . Regulation of p53 localization and transcription by the HECT domain E3 ligase WWP1. Oncogene 2007; 26: 1477–1483.
Li Y, Zhou Z, Chen C . WW domain-containing E3 ubiquitin protein ligase 1 targets p63 transcription factor for ubiquitin-mediated proteasomal degradation and regulates apoptosis. Cell Death Differ 2008; 15: 1941–1951.
Gumienny TL, Lambie E, Hartwieg E, Horvitz HR, Hengartner MO . Genetic control of programmed cell death in the Caenorhabditis elegans hermaphrodite germline. Development 1999; 126: 1011–1022.
Hedgecock EM, Sulston JE, Thomson JN . Mutations affecting programmed cell deaths in the nematode Caenorhabditis elegans. Science 1983; 220: 1277–1279.
van Haaften G, Romeijn R, Pothof J, Koole W, Mullenders LH, Pastink A et al. Identification of conserved pathways of DNA-damage response and radiation protection by genome-wide RNAi. Curr Biol 2006; 16: 1344–1350.
Parsons JL, Tait PS, Finch D, Dianova II, Edelmann MJ, Khoronenkova SV et al. Ubiquitin ligase ARF-BP1/Mule modulates base excision repair. EMBO J 2009; 28: 3207–3215.
Bailly AP, Freeman A, Hall J, Declais AC, Alpi A, Lilley DM et al. The Caenorhabditis elegans homolog of Gen1/Yen1 resolvases links DNA damage signaling to DNA double-strand break repair. PLoS Genet 2010; 6: e1001025.
Alpi A, Pasierbek P, Gartner A, Loidl J . Genetic and cytological characterization of the recombination protein RAD-51 in Caenorhabditis elegans. Chromosoma 2003; 112: 6–16.
Martin JS, Winkelmann N, Petalcorin MI, McIlwraith MJ, Boulton SJ . RAD-51-dependent and -independent roles of a Caenorhabditis elegans BRCA2-related protein during DNA double-strand break repair. Mol Cell Biol 2005; 25: 3127–3139.
Hengartner MO, Horvitz HR . C. elegans cell survival gene ced-9 encodes a functional homolog of the mammalian proto-oncogene bcl-2. Cell 1994; 76: 665–676.
Page BD, Diede SJ, Tenlen JR, Ferguson EL . EEL-1, a Hect E3 ubiquitin ligase, controls asymmetry and persistence of the SKN-1 transcription factor in the early C. elegans embryo. Development 2007; 134: 2303–2314.
Chen D, Kon N, Li M, Zhang W, Qin J, Gu W . ARF-BP1/Mule is a critical mediator of the ARF tumor suppressor. Cell 2005; 121: 1071–1083.
Hall JR, Kow E, Nevis KR, Lu CK, Luce KS, Zhong Q et al. Cdc6 stability is regulated by the Huwe1 ubiquitin ligase after DNA damage. Mol Biol Cell 2007; 18: 3340–3350.
Greiss S, Hall J, Ahmed S, Gartner A . C. elegans SIR-2.1 translocation is linked to a proapoptotic pathway parallel to cep-1/p53 during DNA damage-induced apoptosis. Genes Dev 2008; 22: 2831–2842.
Ito S, Greiss S, Gartner A, Derry WB . Cell-nonautonomous regulation of C. elegans germ cell death by kri-1. Curr Biol 20: 333–338.
Luo J, Shah S, Riabowol K, Mains PE . The Caenorhabditis elegans ing-3 gene regulates ionizing radiation-induced germ-cell apoptosis in a p53-associated pathway. Genetics 2009; 181: 473–482.
Schertel C, Conradt B . C. elegans orthologs of components of the RB tumor suppressor complex have distinct pro-apoptotic functions. Development 2007; 134: 3691–3701.
Derry WB, Bierings R, van Iersel M, Satkunendran T, Reinke V, Rothman JH . Regulation of developmental rate and germ cell proliferation in Caenorhabditis elegans by the p53 gene network. Cell Death Differ 2007; 14: 662–670.
Zahreddine H, Zhang H, Diogon M, Nagamatsu Y, Labouesse M . CRT-1/calreticulin and the E3 ligase EEL-1/HUWE1 control hemidesmosome maturation in C. elegans development. Curr Biol 2010; 20: 322–327.
Hofmann ER, Milstein S, Boulton SJ, Ye M, Hofmann JJ, Stergiou L et al. Caenorhabditis elegans HUS-1 is a DNA damage checkpoint protein required for genome stability and EGL-1-mediated apoptosis. Curr Biol 2002; 12: 1908–1918.
Nehme R, Conradt B . egl-1: a key activator of apoptotic cell death in C. elegans. Oncogene 2008; 27 (Suppl 1): S30–S40.
Akiyama T, Bouillet P, Miyazaki T, Kadono Y, Chikuda H, Chung UI et al. Regulation of osteoclast apoptosis by ubiquitylation of proapoptotic BH3-only Bcl-2 family member Bim. EMBO J 2003; 22: 6653–6664.
Brenner S . The genetics of Caenorhabditis elegans. Genetics 1974; 77: 71–94.
Quevedo C, Kaplan DR, Derry WB . AKT-1 regulates DNA-damage-induced germline apoptosis in C. elegans. Curr Biol 2007; 17: 286–292.
Acknowledgements
We thank the Caenorhabditis Genetics Center, which is funded by the National Institutes of Health Center for Research Resources for providing worm strains. We kindly thank H.R Horvitz (Massachusetts Institute of Technology) for providing the ced-9(n2812)/qC1 strain, B. Conradt and Stephane Rolland (Dartmouth University) for CED-9 antibody and assistance with the western blot protocol. This work was supported by a Canadian Institutes of Health Research award to W.B.D., Natural Sciences and Engineering Research Council of Canada Graduate Scholarship to A.R., and a Canadian Institutes of Health Strategic Training Fellow in the Excellence in Radiation Research for the 21st Century Program to M.L. We are grateful to A. Perrin and S. Ito for helpful input on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by JP Medema
Supplementary Information accompanies the paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article
Cite this article
Ross, A., Li, M., Yu, B. et al. The EEL-1 ubiquitin ligase promotes DNA damage-induced germ cell apoptosis in C. elegans. Cell Death Differ 18, 1140–1149 (2011). https://doi.org/10.1038/cdd.2010.180
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2010.180
Keywords
This article is cited by
-
BRAP-2 promotes DNA damage induced germline apoptosis in C. elegans through the regulation of SKN-1 and AKT-1
Cell Death & Differentiation (2018)
-
Pro-crossover factors regulate damage-dependent apoptosis in the Caenorhabditis elegans germ line
Cell Death & Differentiation (2013)
-
Noncanonical control of C. elegans germline apoptosis by the insulin/IGF-1 and Ras/MAPK signaling pathways
Cell Death & Differentiation (2013)

{kind=link}