
Abstract
Apoptosis is an important physiological process that promotes tissue homeostasis by eliminating unnecessary or malfunctioning cells. Abnormality in this process contributes to tumorigenesis, as well as the resistance to cancer treatment by radiation and chemotherapy. Restoration of normal apoptosis would not only promote cancer cell death and halt tumor progression, but also increase the response to many current cancer therapies. Although apoptosis induction is an important principle of currently used radiation and chemotherapy treatment, uncovering the mechanisms that govern this process, and which are lost during transformation, represents an important direction for realizing improved therapies for the future. This article first briefly reviews aspects of current discovery strategies for new anticancer therapeutics based on intervening in cell death pathways, and then discusses in more detail several cancer-relevant death pathways, which are disabled during transformation and which can be targeted therapeutically. These include anoikis/cell adhesion; energy metabolism and the unfolded protein response. Finally, we introduce a new concept, which utilizes cancer-specific apoptosis induced by oncolytic viruses. The discussion of these topics involves novel targets, compounds and virotherapy.
Similar content being viewed by others

Death Pathways and Novel Cancer Therapies
Cell death and cancer therapy
Escaping apoptotic cell death resulting in unregulated cell proliferation and eventual tumor development is considered one of the hallmarks of oncogenic cell transformation. Cancer cell resistance to radiation and chemotherapy, two apoptosis-inducing treatments, usually occurs as a result of genetic defects in apoptosis signaling pathways and tumor microenvironments where apoptosis is suppressed. Apoptosis is characterized by certain morphological changes, for example, membrane blebbing, nuclear chromatin condensation, DNA fragmentation and apoptotic body formation, and is controlled precisely by a set of well-characterized signaling pathways, which can be induced by either external or internal stimuli.1 Extracellular stimuli are the specific ligands that bind to the death receptors (DRs) on the cell surface and lead to cell death. Intracellular apoptotic stimuli result from stress conditions, for example, exposure to genotoxic agents, detachment, hypoxia or nutrient deprivation, which induces specific intracellular signals leading to apoptosis or even necrosis. The central cellular organelle for intrinsic apoptosis is the mitochondrion, controlling cell fate to either necrosis or apoptosis. Both apoptotic pathways converge downstream with the activation of caspases 3 and 7 to irreversibly commit cells to apoptosis. Present cancer drugs are usually apoptosis inducers, mainly via intrinsic pathways. Future goals involve achieving greater specificity for cancer cells in attacking these pathways. Necrosis is now also recognized as a programmed cell death mode but, in contrast to apoptosis, operating through less clearly defined pathways. Although the ultimate role of necrosis in cancer therapy is less clear, the relationship between the two modes of death appears to be interchangeable depending on the physiological state of cells. Given these observations, it is even more important to pursue a greater understanding of cell death pathways in order to achieve the goal of improved cancer treatments.
Death receptor-induced apoptosis
The external signals, tumor necrosis factor-α (TNF-α), FasL and TRAIL (TNF-related apoptosis-inducing ligand), are well-known ligands for the DRs located on the cell surface, and their binding instantly transduces death signals into cells. Usually, these extrinsic apoptotic signals lead to apoptosis via caspase 8 directly or a mitochondria-mediated pathway (see below). Both tumor necrosis factor receptor and Fas pathways are important components of normal immune functions. Although their activation could also potentially eliminate proliferating cancer cells, the toxic side effects on normal cells limit the development of such therapies. In fact, soluble TNF-α and Fas administered systemically were shown to cause severe toxicity, and TNF-α has only been approved for the combination treatment by isolated limb perfusion of soft tissue sarcoma.2
However, recent studies on the third member of the DR family, namely TRAIL receptors (TNF-related apoptosis-inducing ligand receptor (TRAIL-R)1, 2, 3, 4), have generated exciting opportunities for cancer cell-specific treatment. TRAIL-R1 or DR4, and TRAIL-R2 or DR5 are best known for their involvement in apoptosis. TRAIL binds to its receptor DR4 or DR5 and induces apoptosis via the caspase 8 pathway, similar to Fas. Preclinical studies have shown that TRAIL selectively induces apoptosis in several types of tumor cells but not in normal cells. The mechanism of such cancer-selective apoptosis is not fully understood as TRAIL receptors are constitutively expressed in many normal cells, although two decoy TRAIL receptors (DcR1 and DcR2) exist on the cell surface and can bind to TRAIL without induction of downstream apoptosis signals. The differential expression of these decoy receptors in cancer cells versus normal cells might contribute to a selective induction of apoptosis mediated by TRAIL. Currently, there has been considerable effort directed toward developing anticancer therapies targeting the TRAIL-induced apoptotic pathway.3, 4 For example, lexatumumab, a human agonistic monoclonal antibody to TRAIL receptor, just entered phase II clinical trials.5 Recombinant TRAIL is also being tested in a phase I clinical trial.6
In addition to TRAIL or TRAIL antibodies, agents which enhance TRAIL-mediated signals could also be considered as a therapy for cancers resistant to TRAIL but yet apoptosis competent. The discovery of agents or gene targets (apoptosis suppressors) can be illustrated by the following experimental scheme. First, the cancer cell lines resistant to TRAIL-mediated apoptosis, for example, A2058 melanoma and AsPC1 pancreatic cancer cell lines, used as experimental models for phenotypic selection or screening,7 can be sensitized to TRAIL-induced apoptosis in the presence of a candidate agent, or when the apoptotic repressor is silenced. Apoptosis activation triggered by TRAIL should produce a rapid and synchronous apoptotic response that is favorable for detection and isolation of apoptotic cells by simple apoptosis readouts (see below).7 In the case of discovery of apoptotic repressor targets, combinatorial gene inactivations are usually performed, in which RNAi/ribozyme libraries can be introduced to the TRAIL-resistant cancer cells followed by TRAIL treatment.7, 8 The siRNA/ribozyme sensitizing these cancer cells to the TRAIL-induced apoptosis can be identified leading to the discovery of their targeted apoptotic suppressors. If pooled RNAi or ribozyme libraries are introduced into the cell populations, the apoptotic cells can be detected in or isolated from the nonapoptotic cells by FACS (fluorescence-activated cell sorting). Thus, the genes whose silencing sensitizes cancer cells to TRAIL-induced apoptosis can be identified from the isolated apoptotic cell population as demonstrated in the identification of IGFBP3 as an apoptosis-inducing gene target.7
Intrinsic (mitochondrial) death pathways
The mitochondrion is a central organelle controlling cell death
Mitochondria are the major energy production organelles of cells and have also long been recognized as central players in cell death, including apoptosis and necrosis. These two death modes can interchange depending on factors that are not completely understood, although recent studies have implicated that certain physiological states, for example, intracellular energy store are involved. Mitochondria sense apoptotic signals for example, DNA damage, ischemia and oxidative stress, and initiate cell death by causing permeability changes in the mitochondrial outer membrane (OM). Mitochondrial membrane permeabilization (MMP) leads to the release of apoptogenic proteins for example, cytochrome c (Cyt c) from the intermembrane space (IMS) of the mitochondria into cytosol. Cyt c together with apoptosis protease-activating factor 1 and pro-caspase 9 form a complex that promotes caspase 9 activation and triggers the activation of the downstream caspases, leading to apoptosis. MMP is regarded as a ‘point of no return’ in the apoptotic pathway. Although OM permeabilization can be monitored by immunoblot detection of IMS proteins (e.g., Cyt c, AIF and so on.) in cytosol or nucleus, mitochondrial inner membrane (IM) permeabilization is usually detected by mitochondrial membrane potential (ΔΨm) changes. Mitochondrial morphologic changes (e.g., fusion, fission and ultrastructural changes including cristae remodeling) are specifically related to mitochondrial functional change in response to environmental influences, for example, stress signaling. Mitochondrial fission is a sign of incipient apoptosis.
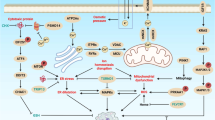
Strategies targeting mitochondria for cancer therapy
Impaired MMP have been associated with cancers and cancer drug resistance, and can result from neoplastic genetic mutations affecting MMP regulators or structural proteins of the mitochondrial permeability transition pore complex (PTPC). Several approaches targeting mitochondria for cancer therapy have been taken (Figure 1). These include those that directly target the IM, Bcl-2 family proteins, PTPC and respiration complexes. At present, more than 20 compounds targeting mitochondria to prompt cancer cell apoptosis are being tested. Several small molecules elicit cancer cell death by targeting mitochondria via direct or indirect interactions with PTPC.9 Betulinic acid (BA) has been shown to selectively kill tumor cells and cause MMP that can be blocked by cyclosporine A, which binds to mitochondrial cyclophilin and suppresses MMP, suggesting action through PTPC. BA is also being tested clinically as a potential cancer therapeutic.10 Arsenite, or other oxidizing agents that deplete glutathione, can antagonize Bcl-2, permeabilize the mitochondrial membrane and result in cancer cell death. It is now a widely used treatment for promyelomonocytic leukemia.11 Lonidamine induces permeabilization of mitochondrial membranes and is also under clinical evaluation (glioma phase II).12

Cancer cell energy metabolism and potential drug intervention. High-energy demand by cancer cells are met via glycolysis (preferentially in many tumors with poor prognosis) and mitochondria oxidative phosphorylation. Different dependence on either of these two processes (i.e., glycolic tumors versus oxidative phosphorylation tumors) would likely respond to treatment differently. Inhibition of energy generation via blocking glycolysis (e.g., 2-deoxyglucose (2DG)) or disturbing the function of mitochondria respiratory chain (e.g., antimycin A, oligomycin) are being considered as new approaches against cancers via targeting energy production machineries
It has been reported that transformed cancer cells or tumors seem to have higher ΔΨm.13, 14, 15 Disruption of ΔΨm can perturb ionic and volumetric homeostasis, and impact mitochondrial functions such as Ca++ signaling and oxidative phosphorylation (OXPHOS). Assays based on mitochondrial dye visualization via flow cytometry, microscopy or UV or fluorescence detection (even by plate reader for high throughput screening (HTS)) have been described to discover agents that affect ΔΨm.16, 17 Using these approaches, a group of structurally related compounds has been identified that cause apoptosis. These compounds, for example, Rhodamine 123, are characterized by delocalized positive charge or delocalized lipophilic cations (DLCs). DLCs were found to target mitochondria, which were driven to a negative ΔΨm.18 F16 is a compound with such structural characteristics and will preferentially accumulate in cancer cell mitochondria, and it also interacts with PTPC. This causes depolarization of IM and results in selective apoptosis of cancer cells and antitumor activity. This type of mitochondriotoxic agent may represent a new anticancer strategy with less toxicity.
Strategies targeting Bcl-2 family proteins
Among the molecules involved in mitochondria-induced cell death, Bcl-2 family proteins have drawn the most attention as potential targets for anticancer therapy. The Bcl-2 family is composed of proapoptotic (e.g., Bax and Bak containing BH1–3 domains (Bcl-2 homology domain) and Bik, Bid, Bad, Noxa, Puma containing only BH3 domain) and antiapoptotic members (e.g., Bcl-2, Bcl-xL, Bcl-w, A1, Mcl-1 and Bcl-B containing BH1-3 domains). The antiapoptotic Bcl-2 family proteins antagonize Bax and Bak's function. Upon apoptotic stimulation, the BH3-only proteins insert their BH3 domain (an α-helix) into a hydrophobic groove of the antiapoptotic Bcl-2 proteins, resulting in the release of Bax and Bak. Oligomerization of Bax and Bak would then affect mitochondrial integrity, promote the release of Cyt c and induce apoptosis.
Bcl-2 and Bcl-xL are overexpressed in many human cancers, and are also believed to be involved in drug resistance. Suppressing their functions or expression promotes tumor cell death and sensitizes tumor cell to radiation or chemotherapy. Novel agents downregulating Bcl-2 are being tested in preclinical studies and clinical trials for cancer treatment. These include: the combination treatment of Genasense (Genta Inc., antisense oligonucleotides targeting Bcl-2, also called G3139, or oblimersen) and dacarbazine for treating melanoma; SPC2996, an LNA-based RNA antagonist of Bcl-2 by Santaris Pharma for chronic lymphocytic leukemia (in phase I/II clinic trial); ABT-737, a small-molecule BH3 mimics targeting Bcl-2, Bcl-xL and Bclw;19 and A-385358 (Abbott Lab),19 a small-molecule inhibitors of Bcl-xL with promising in-vivo results in several animal cancer models.
Strategies targeting IAPs
As described above, MMP-mediated activation of caspases eventually leads to apoptosis. Caspase activation is tightly controlled by the inhibitors of apoptosis proteins (IAPs), a family of structurally related proteins. Eight members have been identified in mammals, including neuronal apoptosis inhibitory protein (also BIR-containing gene 1/BIRC1; X-linked inhibitor of apoptosis (XIAP/MIHA/hILP/BIRC4/ILP-1); cellular IAP1/Human IAP2 (c-IAP1/HIAP2/MIHB/BIRC2); cellular IAP2/Human IAP1 (c-IAP2/HIAP1/MIHC/API2/BIRC3); testis-specific IAP (Ts-IAP//hILP2/BIRC8/ILP-2); BIR-containing ubiquitin conjugating enzyme (BRUCE/Apopllon/BIRC6); survivin (TIAP/BIRC5) and Livin (KIAP/ML-IAP/BIRC7).20 IAPs are structurally similar. They contain one or more 70–80 amino-acid Baculovirus IAP repeat (BIR) domains characterized with C(X)2C(X)6W(X)3D(X)5H(X)6 core sequence that is required for interaction with caspases. Upregulation of IAP proteins has been observed in many cancers, which contributes to resistance to apoptosis. Targeting IAPs promotes cancer cell apoptosis via activation of caspases, or increases susceptibility to chemotherapy agents, which has thus been explored for cancer therapeutics.
Survivin is overexpressed in many tumors (e.g., neuroblastomas, pancreatic cancer, prostate cancer, gastric cancer, colorectal cancer, hepatoma, breast cancer, lung cancer, bladder cancer, melanomas, B-cell lymphomas and esophageal cancers), whereas absent in most adult tissues. Its expression is also correlated with poor prognosis, increased rates of treatment failure and relapse. The differential expression offers treatment selectivity. Antisense oligodeoxynucleotides (AS-ODNs) targeting survivin induce apoptosis, sensitize NSCL cells to etoposide, and are currently under clinical evaluation (phase I, Eli Lilly).21 Cancer vaccines targeting tumor-specific survivin are also currently under clinical evaluations (phase I) using survivin peptides or survivin-directed autologous cytotoxic T lymphocytes.
Although the role in cancer prognosis is less clear, XIAP is the most potent IAP in suppressing caspase activation and apoptosis. XIAP siRNA induces apoptosis or sensitization to etoposide, doxorubicin, TRAIL and taxane, also seen for XIAP-AS-ODNs. XIAP-AS-ODNs combined with either vinorelbine or radiation delayed tumor establishment and caused tumor regression in NSCL xenograft model. XIAP-AS-ODNs (AEG35156) is currently being tested in phase I/II clinical studies.
Smac binds most IAPs via the N-terminal four amino acids of the mature Smac protein (Ala-Val-Pro-Ile) and functions as an IAP inhibitor. The peptide and small-molecule mimics (e.g., peptides with unnatural amino acids, modified oxazoline molecule and polyphenylureas) of the IAP-binding peptides have been explored as potential therapeutics with encouraging efficacy data in preclinical models.
The role of p53 in response to stressors and strategies for directly targeting mutant p53
P53, a classic tumor suppressor, is important in cell-cycle arrest and apoptosis. It protects the integrity of the genome by removing DNA-damaged cells via apoptosis and avoiding mutation accumulation. P53 is also involved in many other stress responses, for example, cell adhesion-regulated cancer cell transformation and metastasis, and energy stress signaling (see below). Approximately 50% of human cancers have mutated p53, rendering them insensitive to genotoxic stressors induced by chemotherapy agents and radiation therapy, and other stressors. Early clinical attempts as well as recent studies have demonstrated the feasibility of restoring p53 activity to sensitize cancer cells to apoptosis induced by chemotherapy.22 Indeed, gene therapy treatment delivering wild-type p53 via a replication-defective adenoviral vector into a cancer with mutated p53 was used to sensitize tumors to radiation therapy. The combination treatment of head and neck cancer via this wild-type p53 delivery and radiation has demonstrated clinical efficacy in reducing tumor progression. This treatment has been approved by the Chinese State Food and Drug Administration, the first gene therapy protocol gaining regulatory approval in the world. In addition, small-molecule drugs for example, PRIMA-1 and CP-31398, have also been shown to halt tumor growth by increasing p53 activity. The regained p53 function caused by PRIMA-1 is likely achieved through partial restoration of the configuration of the misfolded p53, and is thus suitable for tumors with mutated p53.23 In contrast, a compound called nutlins increases cellular p53 activity by disrupting the interaction between p53 and its inhibitor MDM2,24 making it suitable for treating tumors expressing low levels of p53. This compound is proven effective in reducing tumor progression in animal models.
The strategies targeting DNA methylation
Epigenetic control of gene expression by DNA methylation has a great impact on cell proliferation and differentiation. Hypermethylation of promoter regions results in specific suppression of gene expression, including the expression of tumor suppressors, which could promote cancer development. Conversely, demethylation of DNA may enhance cell apoptosis or reduce cell growth. This concept has been proven by a recently approved anticancer drug decitabine for the treatment of myelodysplastic syndrome. Decitabine (Dacogen; MGI Pharma) is a nucleoside analogue that inhibits DNA methylation.25 It demethylates the p73 promoter and induces reexpression of p73, thus activating the caspase cascade and leading to leukemic myeloid cell death.26 DNA hypermethylation in tumor cells may be involved in resistance to interferon (INF)-induced apoptosis, and inhibition of DNA methylation may also enhance the therapeutic effect of INF. Treatment of cancer cells with specific DNA demethylating nucleoside analogue was shown to augment the effect of INF.27
Cell-based assays for intrinsic apoptosis studies
Cell-based assays can also be devised to screen for new drugs or drug targets involving intrinsic apoptotic pathways to sensitize tumors to existing chemotherapies. The concept of a screening/selection process is essentially the same as for extrinsic apoptosis screening described above, except that cancer cell lines relatively resistant to the intrinsic apoptosis-inducing stimuli (e.g., reduced serum level and DNA-damaging chemotherapy drugs) are used, and a specific intrinsic stimulation is used for synthetic lethality. For example, if an RNAi library is used in the screening, the silenced genes in these apoptotic cells may be identified and can serve as potential drug targets to restore the response of cancer cells to specific chemotherapy; similarly, if compound library screening is used, candidate small-molecule sensitizers for chemotherapy can be identified for potential novel combination therapy.
With regard to the readout of cell death, two basic types of methods have been commonly used in either screening or in confirmation experiments. First, quantitations based on cell survival: staining of DNA (e.g., DAPI and propidium iodide; vital staining of mitochondrial oxidative enzymes (e.g., alamarBlue, WST-1 and so on.) and vital readout of intracellular ATP levels. Second, direct measurements of apoptosis based on apoptotic cellular and biochemical phenotypes: caspase activity (usually caspase 3, 7);28 TUNEL activity by ELISA and FACS (quantify DNA fragmentation); direct visualization of DNA fragmentation by gel electrophoresis or annexin V staining via FACS. Many of these assays have been used for drug screening and commercial kits for screening are available. There are also other biochemical, morphological and cell biological assays that can be used in conjunction with the above approaches to fully characterize the cellular fates of cancer cells, including cell-cycle analysis, apoptotic cell morphology, Trypan blue staining, release of LDH.
The above discussion pertains to the relationship of apoptosis to cancer therapy in general terms. However, the ability to intervene in apoptotic pathways in a manner, which is specific to cancer cell survival and growth is of critical importance to deriving novel anticancer agents with greater efficacy and safety than existing agents. Therefore, we discuss in greater detail recent discovery efforts, which target three selected pathways that are highly relevant to cancer cell and solid-tumor survival. Examples of the discovery methodology used in cell-based assays and experimental models are also discussed.
Cancer Drug (Target) Discovery in Cell Adhesion-Anoikis Pathways
Anchorage-independent growth, cell adhesion and anoikis
Anchorage-independent growth is a hallmark of cell transformation and is important in cancer cell invasion and metastasis. Normal cells survive and proliferate by adhering to extracellular matrix (ECM), via ECM-dependent inhibition of apoptosis (Figure 2). These nontransformed cells respond to detachment from ECM by disruption of the actin skeleton (amorphosis), leading to cell rounding and the triggering of anoikis (apoptosis) via activation of proapoptotic Bcl-2 family proteins and the mitochondria-mediated apoptotic pathway (see above). Anchorage-independent growth of tumor cells is largely attributed to reduced anoikis through suppression of these pathways. Anoikis or amorphosis is also an important checkpoint for cancer cell metastasis, because cancer cells need to overcome loss of adhesion during the metastasis process (Figure 2).29 In addition, cell adhesion is also important in drug resistance, which has been demonstrated via Bcl-xL and Akt antiapoptotic pathways.13 As anoikis resistance is a rather unique property of cancer cells, adhesion and/or anoikis pathways are considered useful targets for cancer intervention with improved specificity over conventional chemotherapies. In fact, interference of cell adhesion via targeting cell–ECM or cell–cell interaction is currently being evaluated in clinical trials for cancer therapy.

Interaction of cancer cells with extracellular matrix (ECM) is important for cell proliferation and survival
Receptors mediating cell adhesion are mainly composed of four families of cell-surface proteins: integrins, cadherins, selectins and immunoglobulins. Integrins have been found to play important roles in carcinogenesis, and therefore are the most intensively studied for cancer treatment.30 Integrins are transmembrane heterodimers of α and β subunits, and the combination of different isoforms of each subunit can form a total of 24 integrin dimers with different ligand-binding specificities. The interaction of integrins with ECM proteins such as fibronectin triggers a series of cellular signaling events to regulate cancer cell survival, proliferation and migration. Intervention in integrin-mediated cell adhesion can be achieved by either targeting the receptor itself or by targeting the intracellular signaling molecules involved in integrin pathways. Several drug approaches are currently being evaluated for potential clinical application based on inhibition of cell-adhesion pathways: specific antibody for αvβ3 integrins, small-molecule antagonists of integrin receptor and arginine-glycine-aspartic acid (RGD) peptides/peptidemimetics. RGD is found in several important integrin ligands belonging to ECM proteins, and the peptides containing RGD can potentially bind to integrin and interfering with cell adhesion.
Intracellular signaling proteins downstream of integrins can also be targeted. ErK and PI3K are well-known kinase targets for cancer therapy as they are not only important for cancer cell proliferation, but also play crucial roles in integrin-induced cell adhesion. Inhibitors of these enzymes are currently being evaluated in the clinic. Integrin-linked kinase (ILK) represents a novel integrin pathway, which is emerging as a promising cancer target.31, 32 ILK is a unique intracellular adapter that links the cell-adhesion receptors, integrins and growth factors to the actin skeleton and cellular signaling pathways. ILK expression is found to be elevated in different cancers, and inhibition of ILK reduces cancer progression.
Cadherin signaling-mediated cell–cell interaction is also found to be an important player in cancer cell metastasis and invasion. In particular, E-cadherin is believed to have an inhibitory function on cancer cell invasion due to the fact that loss of E-cadherin is associated with epithelial cancer metastasis and malignancy. However, increasing evidence also suggests a ‘cadherin switching’ mechanism in certain metastatic cancers, in which normal functional E-cadherin is maintained, but N-cadherin levels are upregulated. These studies imply that N-cadherin could be an attractive potential cancer target for prevention or inhibition of tumor metastasis.33
Cell-based assays for anchorage-dependent and -independent growth
One of the requirements for drug discovery based on these pathways is a robust phenotype assay. First, conventional cell growth assays allow cell growth in liquid culture where cells are attached to the vessels. This two-dimensional monolayer culture mimics cell attachment to ECM (cell–ECM adhesion) and measures anchorage-dependent cell growth. Various formats and readouts, for example, 96-well and colony formation formats, are commonly used to screen and/or confirm targets and agents for inhibition of cell growth.34, 35 Second, by detaching and reattaching cells to the ECM (e.g., fibronectin), one can also directly assess intergrin-mediated adhesion properties of cancer cells under different treatments by directly quantitating the attached versus detached cells using common cell quantitation methods.36, 37 Third, growth of multicellular aggregates in three dimensions (spheroid) has also been devised, where cancer cells attach to each other via cadherin-dependent junctions (cell–cell adhesion) and more closely mimic tumor growth in vivo than a two-dimensional system. This spheroidal growth is usually more resistant to apoptosis induced by chemotherapy than the same cells grown under cell–ECM conditions,38 as the interior of spheroids may mimic the hypoxic and hypoglycemic microenvironment seen in solid-tumor interior.39 Therefore, spheroid growth has been used to test the effects of agents on cadherin-mediated adhesion and signaling, and as a predictor of inhibition of solid-tumor growth.39 Fourth, soft agar growth is a classic anchorage-independent growth assay where cancer cells proliferate in semisolid soft agar and form colonies. The size and number of colonies can be used as a measurement of transformation (anchorage-independent growth) potential. Soft agar growth is viewed as the closest mimic of in-vivo tumorigenicity among the in-vitro assays. Soft agar colony-based testing can be automated.35 A 96-well soft agar assay can also be readily applied to HTS of cancer targets or anticancer agents.34 Fifth, anoikis can also be directly assessed for cancer cells by direct and quantitative measurement of apoptosis of cells after detachment.40
HeLa/HeLaHF transformation cell pair system
Another important tool for discovering targets involved in anoikis-related pathways is a robust and relevant experimental cancer system. HeLa, a cervical adenocarcinoma cell line, is a classic experimental cancer cell line broadly used in oncology research over the past half century. It shows strong transformed phenotypes, including anchorage-independent growth in vitro and tumorigenesis in vivo. HeLaHF is a revertant variant isolated from HeLa cells following exposure to the mutagen EMS by Zarbl and colleagues.41 HeLaHF demonstrates a ‘nontransformed’ phenotype exhibited by the appearance of flat and nonrefractile morphology, significantly decreased growth in soft agar, and loss of in-vivo tumorigenicity. We have also confirmed that the phenotypic difference between the HeLa and HeLaHF cell pair in transformation is largely due to their difference in anoikis levels.42 Cell fusion studies indicated that the transformation reversion likely results from the activation of tumor suppressor(s).41 As HeLa and HeLaHF constitute an ‘isogenic cell pair’ with distinct and readily measurable transformation phenotypes, the pair is an ideal system for identification of genes involved in HeLa-cell transformation (anchorage-independent growth). Many of the genes that are expressed differentially in this cell pair could be interesting candidate targets for cancer therapeutics, if they play similar roles in other cancer systems.
Discovery of genes involved in HeLaHF cell anoikis by functional genomic approaches
The role of the p53 pathway in anoikis in HeLaHF
Expression profiling analysis coupled with functional studies based on either loss-of-function (LOF) or gain-of-function can be used to identify new gene targets and pathways involved in anoikis-related cellular transformation of the HeLa cell system. Zarbl and colleagues noticed that p53 proteins were stabilized in HeLaHF cells and that several genes in the p53-mediated pathway were also differentially expressed between the HeLaHF and HeLa cell pairs as detected by expression profiling. These include IGFBP3 and DKK1, which are overexpressed in the revertant HeLaHF cells. When IGFBP3 and DKK1 transgenes were overexpressed in transformed HeLa cells, these researchers observed that the tumorigenicity was effectively suppressed, suggesting that the p53 pathway is causally involved in HeLa transformation. We provided further evidence through p53 silencing experiments that p53 downregulation reduced anoikis in HeLaHF cells, and increased anchorage-independent growth by10-fold.42 The involvement of p53 in anchorage-independent growth is consistent with its known role in cell–ECM interactions such as integrin signaling. Integrin-mediated survival signaling is only effective in p53-deficient cancer cells, and cancer cells with wild-type p53 are able to undergo apoptosis in response to this cell–ECM interaction-initiated signal.43, 44 Thus, for cancer cells to survive and grow in an anchorage-independent manner, inactivation of p53 is important to prevent cell anoikis. The role of p53 in anchorage-independent growth is not only seen in HeLa/HelaHF cell pairs, but also demonstrated in other cell systems, including HCT116 colon cancer cells and the MCF-7 breast cancer cell line35, 43, 44 (Li et al., unpublished data), where inactivation of p53 increased the proliferation rate for anchorage-independent growth.35
However, p53 apparently only contributes partially to the enhanced anoikis in HeLaHF cells based on the two observations. First, the near-complete silencing of p53 was insufficient to reactivate the full transformation potential of HeLaHF to the levels of HeLa cells, suggesting that additional factors must be involved in HeLaHF reversion. Second, the gene expression changes induced by p53 silencing in HeLaHF cells only constitute a small portion of the gene pools that are differentially expressed between HeLa and HeLaHF cell pairs. In this regard, other genes that are also involved in the transformation reversion have been identified from the comparative gene-profiling data. Using a similar approach, a novel gene putative HeLa tumor suppressor that promotes the transformation reversion from HeLa to HeLaHF via a p53-independent pathway was also identified.42
Pathways that are important in maintaining HeLa-cell transformation
The identification of tumor suppressor genes that are overexpressed in HeLaHF cells may elucidate the mechanisms underlying the reduced tumorigenecity of HelaHF. However, the discovery of ‘oncogenes’, rather than tumor suppressor genes, can be more interesting as they are more likely to be useful cancer targets. Among the genes upregulated in HeLa cells were the well-known oncogenes, for example, polo-like kinase (PLK), survivin, chemokine receptor CXCR4 (also see below). The causal roles of PLK45 and CXCR4 (Ke et al., unpublished data) in HeLa-cell transformation have been confirmed, which are consistent with previous published studies of these genes using different experimental systems.
Interestingly, three members of the Nur factor subfamily of nuclear receptors, NR4A1, NR4A2 and NR4A3, are all upregulated in transformed HeLa cells as compared to nontransformed HeLaHF cells, suggesting potential oncogenic properties. RNAi-mediated silencing of any of these members in HeLa cells promotes apoptosis or enhanced anoikis (reduced anchorage-independent growth), as well as reduced tumorigenicity40, 46 (Ke et al., unpublished data), consistent with report by other researchers.47 It is also important to point out that the opposite effect exerted by this family of receptors, a proapoptotic role, has also been extensively reported and reviewed.48 In particular, a recent report has also linked the proapoptotic property to human leukemia.49 Nuclear receptor family proteins are considered to be highly targetable by small molecules. As a matter of fact, it was recently reported that modulators for this family of receptors have been identified, suggesting the potential of targeting these receptors for novel therapies.50
Anoikis is a typical response to cell detachment, so it is not surprising that proteins functioning in cell-adhesion pathways are involved in HeLaHF reversions. Indeed, the differential expression of genes in the HeLa and HeLaHF cell pairs demonstrates that the expression of the known adhesion signal genes is found to be upregulated in HeLaHF, including tyrosine kinases such as Fyn and Fak (which are also proto-oncogenes) as well as integrins (Figure 2). RNAi experiments confirmed the causal role of Fyn in HeLa42 and A205851 transformation. The expression profiling also identified an orphan G-protein-coupled receptor belonging to the adhesion GPCR subfamily, GPR56. The mutations in GPR56 have been associated with bilateral frontoparietal polymicrogyria, a rare neurological disease that affects brain development,52 which results from the lack of surface expression the mutant proteins.53, 54 RNAi-mediated silencing of GPR56 resulted in increased anoikis and reduced anchorage-independent growth of HeLa and other cancer cells, whereas cDNA overexpression resulted in increased focus formation in the mouse fibroblast NIH3T3 cell line.37 More significantly, when GPR56 silencing was induced in vivo in several xenograft tumor models, significant tumor responses (including tumor regression) were observed, suggesting the potential of targeting GPR56 in cancer therapies.
Silencing of GPR56 in the A2058 melanoma cell line affected the expression of a number of downstream genes. Of particular interest are those genes known to be involved in the integrin-mediated signaling and cell-adhesion pathways, such as Fak, Fyn and integrins.37 The role of GPR56 in cancer-cell adhesion was further confirmed by the observation that GPR56 silencing also reduced cell adhesion to the ECM, which is consistent with the observed increase in anoikis and reduction in anchorage-independent growth phenotypes (Figure 2). This involvement of GPR56 is consistent with an earlier report.55 The oncogenic potential and the apparent absence of physiological defects in adult human tissues lacking GPR56, along with the ‘druggable’ nature of GPCR family proteins, make GPR56 an attractive drug target. It is important also to point out that GPR56 was also recently implicated in cell adhesion but with an opposite effect to that observed in several different experimental systems.56
Besides gene expression profiling-based discovery schemes, combinatorial gene silencing has also become a common approach used to identify targets. Essentially, libraries of either ribozyme,7, 57, 58 RNAi8, 59, 60 or even cDNA, can be utilized for combinatorial gene silencing using the nontransformed HeLaHF cells to discover gene targets involved in HeLa-cell transformation/anoikis resistance. As an example the human ortholog of the Drosophila ppan gene and integral membrane protein 2B (a protein implicated in mitochondrial mediated apoptosis7, 58), where many aspects of this technology have been previously described and reviewed.
Treatment Strategies Targeting Energy Metabolism Pathways of Cancer Cells
Elevated energy demand, particularly met by increased glycolysis, is a hallmark of transformation
Tumors have significant higher demand for energy than normal tissues in order to survive and grow. Glycolysis, the anaerobic breakdown of glucose into ATP, is a preferred method to meet the demand for high rates of ATP production in cancer cells, in contrast to normal cells that mainly rely on respiration, or OXPHOS. It has recently been found that mitochondrial OXPHOS was inversely correlated to glycolytic activity, as demonstrated by positron emission tomography (PET).61 Significantly enhanced anaerobic glycolysis, or the Warburg effect, is also considered an important characteristic of many cancer cells, reflected by both the increased activities of the glycolytic enzymes (e.g., hexokinase II (HK II), PFK I, pyruvate kinase) and the glucose transporter (e.g., Glut-1). This is exploited for tumor diagnosis by imaging the uptake of radioactive fluorine-18 deoxy-D-glucose (also a glucose analogue) using PET. The enhanced uptake of FDG by tumors as shown by PET, in contrast to normal tissues, not only quantifies and confirms the different degrees of glycolysis increase in different tumors (‘glycolytic’ versus ‘OXPHOS’ tumors), but also increasingly serves to functionally monitor tumor progression and tumor response to therapy, as compared to the traditional imaging techniques for example, computed tomography (CT scan), which are based solely on anatomical as opposed to functional readouts. Although it is still unclear whether changes in energy metabolism are the cause or the result of cell transformation, targeting the underlying biochemical pathways represents a new opportunity for cancer intervention. High glycolytic dependency has been associated with poorer prognosis and poorer responses to treatment. Furthermore, tumors with different energy metabolic characteristics, either glycolytic or OXPHOS, likely respond differently to treatments targeting energy metabolism. Using PET/CT could potentially guide such treatments as a personalized approach, although this concept remains to be tested in the clinic.
Strategies targeting cancer cell energy metabolisms
Strategies directly targeting glycolysis
Glycolysis occurring in the cytosol is one of the two main sources of cellular ATP production. Targeting glycolysis directly is therefore a straightforward approach to disrupting the energy metabolism of tumor cells for antitumor activity. There are several approaches that have been taken, including inhibiting glycolytic enzymes and glucose transporters and induction of release of HK II from the mitochondrial receptor (VPAC) (Figure 1). The compound 2-deoxyglucose (2DG) is an antimetabolite and a glycolysis inhibitor that is also preferentially taken up by tumors, similar to the uptake of glucose. 2DG has been shown to cause cancer cell apoptosis in vitro and to also further upregulate Glut-1,62 mimicking glucose deprivation. However although it alone seems to have very little antitumor activity, its combination with other chemotherapies, for example, adriamycin and Taxol, demonstrated antitumor activity in animal models,63 and possibly in patients. One of the advantages of 2DG over many other conventional treatments is that it is considered a nontoxic agent and may potentially offer better quality of life than other cytotoxic agents. Clinical studies have demonstrated that it was well tolerated (>200 mg per kg) and seems to have clinical benefits as combination therapies with radiation and chemotherapies.64 A chemotherapy drug conjugated to glucose is also being tested (Threshold Pharmaceuticals Inc.) to take advantage of the increased glucose uptake by tumor for specific targeting of chemotherapy. HK inhibitors, for example, 3-bromopyruvate, have been shown to have impressive activity against liver cancers.65 Phloretin, a Glut-1 inhibitor, was shown to suppress glucose uptake by 60% and sensitize cancer cells to daunorubicin, suggesting yet another antitumor scheme.
Targeting pathways sensitizing cancer cells to glycolytic suppression
An alternative approach to exploit glycolysis inhibition for cancer therapy is to identify agents that can further sensitize cancer cells under glycolytic inhibition. It has been hypothesized that glycolysis inhibition-mediated cytotoxicity is partially mediated by disrupting thiol metabolism and causing metabolic oxidative stress (see below). It was proposed that a strategy of inhibiting hydroperoxide metabolism may sensitize cancer cells to glucose deprivation and 2DG, for example, suppression of GCL.66 It is also interesting to consider that Gleevec (imatinib), a kinase inhibitor, significantly reduces glucose uptake by the treated tumor, implicating a role of glycolysis inhibition in the action of Gleevec.67, 68 Pyrvinium pamoate, an antihelminthic medicine, was recently reported to have selective toxicity for cancer cells under conditions of glucose deprivation in vitro.39 This selectivity was apparently exerted in vivo as reflected in the killing of pancreatic cancer cells within the tumor interior, where lower glucose levels can be expected.
Targeting OXPHOS via proteins that reside on the IM
Another important site of cellular energy production is the mitochondrion. Respiratory chain components are located on the IM, which produces the majority of ATP that cells need. There are five complexes that are responsible for respiration and energy production (I, II, III, IV and ATP synthase (V). Targeting each component of this essential bioenergetic process has been considered for cancer therapy, including the use of antimycin A analogs, which inhibit complex III; rotenone, which targets complex I,69 and oligomycin, which targets ATP synthase or complex V. All of these agents have been shown to have antitumor activity. It is conceivable that combination treatment involving the targeting of glycolysis and mitochondrial energy metabolism would be a potent and synergistic approach. However, the therapeutic margin achieveable using these agents would have to be large enough to avoid toxicities due to inhibition of energy metabolism in normal tissues. As mentioned above, mitochondria also play a key role in regulating cell death. A therapeutic strategy for cancer that could unite the cell death and bioenergetic abnormalities of tumor cells could provide for a novel and powerful treatment.
Strategies for Targeting Stress Responses in Solid Tumors
Hypoxia and hypoglycemia are two important characteristics of solid tumors
Intratumoral heterogeneity is a common characteristic of solid tumors (Figure 3). Poor vascularization results in insufficient perfusion within solid tumors. The interior of solid tumors is usually deprived of oxygen and nutrients, for example, amino acids and glucose. Hypoglycemia is further enhanced by a greater demand for glucose as an energy source by cancer cells. The consequences of these tumor microenvironments include inhibition of glycolysis and OXPHOS that result in insufficient production of ATP and an increase in the production of reactive oxygen species (ROS). These stresses on mitochondria contribute to apoptosis or necrosis of cancer cells inside the solid tumor. This effect is the very basis of antiangiogenic therapy for solid tumors, for example, Avastin (an anti-VEGF monoclonal antibody) (Figure 3). To adapt to these stress conditions, cancer cells in the tumor interior often become dormant via several protective mechanisms, including the unfolded protein response (UPR) and HIF-1α (hypoxia-induced factor-1α) gene upregulation (see below) and HK II, Glut-1,3, VEGF and AM (adrenomedullin) gene upregulation, to counter stress effects and avoid cell death. In addition, the dormant interior is also more resistant to chemotherapies that usually require O2 and active cell division, constituting an important mechanism of treatment resistance and rendering poor therapy outcome of solid tumors. However, understanding the mechanisms of these stress responses could also represent new intervention opportunities.

Heterogenic microenvironment of solid tumors and novel therapeutics targeting the hypoxic/hypoglycemic tumor interiors
HIF-1α, AMPK and mTOR are involved in stress responses in solid tumors
HIF-1α is a transcription factor responsible for upregulating angiogenic factors (e.g., VEGF) to promote vascularization within tumors and upregulating glycolytic enzymes (see above) and glucose transporter (e.g., Glut-1) to increase ATP production, thus countering the stresses. An important response to hypoxia is the activation of HIF1α via stabilization, which subsequently induces downstream gene expression to counter the hypoxic stresses. The activation of HIF-1α has increasingly been recognized as a common consequence of many human neoplastic mutations, and is also considered an important characteristic of solid tumors within the hypoxic loci. Thus, targeting HIF1α becomes yet another tumor specific treatment approach against solid tumors.70
Another key molecule for sensing intracellular energy supply is AMP-activated protein kinase (AMPK).71 It is activated via phosphorylation when the cellular AMP/ATP ratio increases, for example, when there is a lack of energy due to a reduction in levels of glucose, amino acids and oxygen. AMPK downregulates the TSC–mTOR pathways and also activates p53, resulting in cell growth arrest or apoptosis. In fact, AMPK is found to be activated in the hypoxic and hypoglycemic solid-tumor microenvironment,72 suggesting a protective mechanism of AMPK and its importance to tumor survival. However, whether or how regulating AMPK can benefit cancer treatment remains to be seen. Nevertheless, AMPK agonists, for example, the antidiabetic drug TZDs, were found to have anticancer activity in animal models in concert with downregulation of the mTOR pathway, suggesting yet another approach to target energy metabolism-related transformation pathways.73 The mTOR pathway is well-known for its mediation of cells responses to nutrients. Activation of the mTOR pathway promotes cell proliferation and inhibits cell death. Targeting mTOR-mediated energy metabolic pathways has long been investigated as an antitumor strategy. This concept is also supported by the fact that the prototypic mTOR inhibitor rapamycin has demonstrated antitumor activity in certain human cancers. The first mTORinhibitor to be approved for cancer is temsirolimus (Torisel), which was approved in 2007 for end-stage renal cell carcinoma. The pTEN–mTOR pathway is also involved in the growth signaling responses stimulated by extracellular growth factors for example, IGF and EGF, adding yet another dimension to the antitumor potential of mTOR inhibition.74
The UPR prevents apoptosis induced by stress
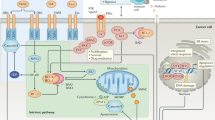
The UPR consists of the signaling networks responding to aberrant protein folding in the endoplasmic reticulum (ER) (Figure 4). It involves the expression of protein chaperones and folding catalysts that maintain the physiological balance between folding demand and capacity of the ER. Hypoxia, hypoglycemia and genotoxic agents (e.g., chemotherapy agents like doxorubicin, etoposide) all induce the UPR, although the underlying mechanisms are yet to be fully elucidated). The UPR functions as a major protective mechanism under ER stress, and is usually a transient event. However, chronic exposure to ER stress induces CHOP expression and increases the production of ROS leading to cell death.75

Endoplasmic reticulum (ER) stress-mediated unfolded protein response (UPR) signal pathways
Glucose-regulated protein 78 (GRP78), an ER chaperone molecule and also known as immunoglobulin heavy chain-binding protein, is a key molecule in the UPR with two functional domains: an N-terminal ATPase domain and a C-terminal polypeptide-binding domain. The binding affinity of GRP78 with polypeptides depends on the association with ATP or ADP at its ATPase domain. The ATP-bound form has a lower affinity for unfolded proteins, whereas the ADP-bound form has higher peptide-binding affinity. Ectopic expression of GRP78 in human gastric carcinoma cells resulted in enhanced resistance to apoptosis induced by celecoxib, a nonsteroidal antiinflammatory drug (Cox-2 inhibitor) that was able to induce apoptosis in cancer cells. Conversely, silencing of GRP78 via RNAi or antisense DNA sensitize cancer cells to cis-platin-induced apoptosis. Three mechanisms have been proposed for the antiapoptotic role of GRP78. First, GRP78, as a major ER chaperone, alleviates the accumulation of unfolded protein in ER and reduces apoptotic signaling. Second, GRP78 regulates ER Ca2+ storage and reduces Ca2+ overloading in mitochondria.76 Third, GRP78 physically binds to caspase 12 and inactivates its activity,76, 77 although the significance of this is controversial as the caspase-12 activity has been found absent in many human cells.
In addition to GRP78, several other key signal molecules in UPR, for example, XBP-1, PERK and ATF4, have also demonstrated their antiapoptotic properties. Cells with a PERK deletion mutation (PERK−/−) were more sensitive to hypoxia-induced cell death than the wild-type cells, correlating with increased caspase 12 activation. Stable expression of dominant-negative PERK in HT29 colorectal carcinoma cells also increases apoptosis.
Evidences for UPR involvement in solid-tumor development
Increasing evidence indicates a critical role for the UPR in tumor development, progression and resistance to chemotherapeutic agents. This involvement is apparently HIF-1 independent. The importance of GRP78 in tumor growth has been supported by several observations. Overexpression of GRP78 has been found in many human tumors, for example, aggressive breast cancer,78, 79 gastric cancer,80 lung cancer,81 hepatocellular carcinoma82 and prostate cancer.83 Furthermore, this upregulation is also correlated to the aggressive malignant phenotype and the poor prognosis post treatments. GRP78 is also found to contribute to the switch of energy metabolism from respiratory to glycolysis in hypoxic- and glucose-deprived solid tumors,84, 85 a hallmark for cancer progression. Jamora et al.86 showed that downregulation of GRP78 resulted in impaired tumor formation, or rapid tumor regression as demonstrated in a mouse fibrosarcoma xenograft model. The PERK-eIF2α pathway also plays a positive role in tumor growth as the transformed cells lacking PERK and eIF2α function form tumors that grow ∼6-fold slower than those with a functional PERK-eIF2α pathway.87 ATF4, a downstream target of PERK, has also been associated with the development of many solid tumors.
Tumor metastasis occurs through a complex series of steps and requires many acquired cancer-specific properties, for example, invasion, migration, angiogenesis and adaptation to adverse host environment (stresses). Emerging evidences also point to the association of GRP78 with tumor metastasis. Increased GRP78 levels are detected in metastatic tumor cells.88, 89 The receptor-recognized form of the proteinase inhibitor α2-macroglobulin (α2M) was found to physically associate with GRP78 and activate Pak-2-dependent signaling. Because activation of Pak-2 promotes migration of cancer cells, this observation suggests that GRP78 may facilitate tumor metastasis through this α2M–PAK-2 pathway.90 Furthermore, the angiogenic cytokines VEGF and IL-8/CXCL8 was found to be responsive to nutrient deprivation (e.g., glucose) and ER stresses.91
Overexpression of the XBP-1 was also observed in colorectal carcinomas and adenomas.92 The direct involvement in solid-tumor growth was first demonstrated from the functional study on Ire1/XBP-1 pathway. Cancer cells containing a homozygous deletion mutation of XBP-1 gene showed impaired survival under hypoxia. These XBP-1−/− cells also failed to form tumors in animals. Experiment using a human tumor xenograft expressing an XBP-1s-luciferase fusion protein demonstrated the expression of spliced active XBP-1s was detected in all tumors including small ones,93 suggesting that the ER stresses persistent throughout tumor growth, and the Ire1/XBP-1 pathway plays a critical role in solid-tumor progression.
Evidence for the role of the UPR in drug resistance of solid tumors
Activation of the UPR, for example, induction of GRPs was found to induce resistance to adriamycin (or doxorubicin). GRP78 can specifically bind and inactivate caspase 7, a downstream inducer of apoptosis activated by many anticancer chemotherapy reagents.94 Examination of a panel of human breast cancer cells revealed that the expression levels of GRP78 positively correlates with their resistance to various drug-treatment regimens, including adriamycin and etoposide.84 In addition, cells expressing a mutant GRP78 deficient in binding to caspase 7 showed increased sensitivity to etoposide-induced killing.
Anticancer agents targeting pre-existing blood vessels or new vessel formation in tumors represent new classes of anticancer therapeutics and show promise in delaying tumor progression. However, this class of agents may also enhance glucose and oxygen deprivation within the solid tumors that could trigger unintentional prosurvival UPR. Using the human breast cancer xenograft model, Dong et al.84 showed that the vascular targeting agent, combretastatin A4P, or the antiangiogenic agent, contortrostatin, induces GRP78 in surviving tumor cells. Suppression of GRP78 by lentiviral vector expression-specific siRNA sensitizes etoposide-induced cell death. Lee and colleagues recently examined the expression of GRP78 from archival tumor specimen from breast cancer patients treated with adriamycin. They found that the degree of GRP78 expression is well associated with the ‘time of recurrence’. This result suggests that GRP78 can be used as a predictor for chemo-responsiveness,95 and an improved clinical efficacy can also be achieved through a combination of the agents targeting UPR with current chemotherapies and/or antiangiogenic agents.
Treatment strategies of solid tumors by targeting the UPR
In light of the evidence of UPR involvement in solid-tumor development and drug resistance, targeting UPR signaling pathways becomes an attractive approach to explore new novel class of cancer therapeutic agents against solid tumors.
Targeting GRP78
The ability to reverse malignant phenotypes of cancer cells by silencing GRP78 via RNAi or antisense suggests targeting GRP78 as a potential new anticancer strategy. Indeed, several agents targeting GRP78 have recently been reported. EGCG ((-)-epigallocatechin gallate), an agent with chemopreventative, anticarcinogenic and antioxidant properties, was found to directly bind to GRP78 at its ATP-binding site and inhibit its ATPase activity.96 The association of EGCG with GRP78 suppresses the formation of the antiapoptotic GRP78-caspase-7 complex and sensitizes etoposide-induced cancer cell death. Versipelostatin, a natural product derived from a bacterium, was initially identified to suppress the activation of ER chaperones for example, GRP78 and GRP94 in response to glucose deprivation and sensitize tumor cell to glucose starvation. Furthermore, versipelostatin can also suppress tumor growth either alone or in combination with cisplatin in human stomach cancer xenograft model.97 GRP78 has been recently found to be localized to the cancer cell surface (and not in normal tissues) in addition to the ER. A synthetic chimeric peptide composed of the GRP78-binding motifs fused with a cell death-inducing sequence inhibited tumor growth in human prostate and breast cancer xenograft mouse models.83 This result demonstrates that GRP78 on tumor cell surfaces can also be directly targeted for anticancer therapy. Interestingly, cell-surface GRP78 was shown to be a receptor for Kringle 5 (K5), an antiangiogenesis inhibitor.98 The high-affinity binding of K5 with GRP78 on proliferating endothelial cells and stressed tumor cells causes apoptosis, which suggests a potential anticancer application of recombinant K5.
A reporter system can be a convenient method to discover agents that downregulate GRP78 as GRP78 is largely regulated at the transcriptional level. Recently, a cell-based system stably expressing the luciferase gene under the control of the GRP78 promoter was described for screening of downregulators of GRP78. A number of compounds that selectively inhibit GRP78 expression under ER stress have been identified, which include verrucosidin, valinomycin and prunustain A. However, the in-vivo antitumor activity and mechanism of action of these compounds remain to be elucidated.
Targeting Ire1/XBP1
The critical role of Ire1/XBP-1 pathway in tumor cell survival and growth prompts the development of two approaches for the Ire1/XBP-1-based therapies: discovering selective inhibitors of Ire1 and screening for novel agents that downregulate XBP-1 expression using a reporter system. Feldman and his colleagues recently identified several compounds that inhibit Ire1-mediated splicing of XBP-1 while their antitumor activities are still being investigated.
Cancer Cell-Specific Apoptosis Induced by Oncolytic Viruses (Virotherapy)
Virotherapy is an exciting new therapeutic approach, which may potentially be more efficacious and less toxic than previous cancer therapies. Virotherapy is largely based on selective replication of certain lytic viruses, either naturally occurring or engineered, in tumor cells versus normal cells, leading to lysis of the tumor cells (oncolytic effect). The inability of these viruses to productively infect normal cells can be rescued by activation/inactivation of specific gene pathways in cancer cells, including Ras activation, p53 inactivation and Akt signaling activation, depending on the specific oncolytic viruse. Virotherapy has now been shown to act through multiple mechanisms, including oncolysis and the induction of tumor-specific apoptosis.99 Some of these oncolytic viruses have been tested in human clinical trials and have demonstrated favorable initial safety and efficacy profiles.100
One mechanism of host defense against lytic viral infection is the induction of premature apoptotic cell death preventing productive infection (spreading). This can occur in several ways, including an INF response and/or innate immune responses. Most lytic viruses are equipped with specific mechanisms for inhibiting infection-aborting apoptosis, including herpes simplex virus (HSV),101 and vaccinia virus. For example, HSV infection initially triggers the apoptosis process but subsequently one of the viral-encoded proteins (ICPs) blocks the actual death of the infected cells in supporting the completion of viral replication.102
Interestingly, deletion of certain of these key viral genes involved in blocking premature apoptosis prevents the virus from spreading (productive infection) in normal nontransformed cells and tissues, but has no impact on the virus' ability to infect cancer cells, thus converting these viruses into ‘oncolytic’ viruses. The recombinant vaccinia virus with deletion of the thymidine kinase gene becomes an oncolytic virus as it replicates in cancer cells but not normal cells. Adenovirus with its transforming gene E1B deleted cannot productively infect normal cells with wild-type p53, but can productively infect cancer cells with LOF of p53.22 The PK (protein kinase) domain of the HSV-2 ICP-10 gene was believed to be important in inactivating the ras signaling pathway of infected cells, which is essential to viral infection. The deletion of the PK domain renders HSV-2 unable to infect normal cells, but able to infect human cancer cells with the activated ras signaling pathway. Thus, recombinant ICP10-PK(-)HSV-2 is a potent oncolytic virus (FusON2).103, 104 In the case of HSV-1, deletion of the γ34.5 gene also disables its infection in nontransformed cells while maintaining its capacity to infect cancer cells.105, 106
Oncolytic viruses specifically kill tumor cells via viral mediated cell lysis, a process called oncolysis. Recent studies demonstrated that certain oncolytic viruses can also specifically induce apoptosis in some cancer cells both in vitro and in vivo, while still managing to lead to productive infection, in contrast to the situations described above. The apoptosis seems also to contribute to the enhanced antitumor effect in vivo.103 One possible explanation is that apoptosis is inhibited at the early stage of viral infection as designed, but reinduced at a later stage when the viruses have already replicated. For instance, Fusogenic HSV-2 oncolytic viruses with deletion of the ICP10 PK domain were able to induce apoptosis in tumor cells.103 HSV-1 oncolytic viruses are also found to induce apoptosis in infected tumor cells.107 Meanwhile, studies have also indicated that apoptosis is not only induced in the infected cells, but also in neighboring uninfected tumor cells, a phenomenon called the bystander effect. This effect can further enhance the apoptosis elicited by oncolytic viruses.103, 107 However, the mechanism for this bystander effect is still not clear.
Certain naturally occurring oncolytic viruses can also kill cancer cells via apoptosis. Newcastle disease virus (NDV) is an oncolytic virus currently under clinical investigation for treatment of glioma, and its oncolytic effect has been attributed to apoptosis, mainly mediated by the mitochondria/caspase-dependent pathways and independent of INF responses. DR-mediated tumor cell apoptosis is a late event of NDV infection, demonstrated by the fact that inhibition of DR suppressed cell apoptosis, whereas it did not affect viral replication.108 In contrast, reovirus, induces apoptosis via both DR- and mitochondria-mediated pathways.109 Thus, although triggering apoptosis is one common way for oncolytic viruses to eliminate cancer cells, the underlying pathways mediated by different viruses may not be the same.
In summary, virotherapy could represent a powerful new treatment modality for cancer in the near future. Cancer cell-specific apoptosis induced by oncolytic viruses, although is just one of the antitumor mechanisms of virotherapy, it is important in the overall antitumor effect by synergistically working with other antitumor mechanisms of oncolytic virotherapy, such as induction of antitumor immunity.
Abbreviations
- ΔΨm:
-
mitochondrial membrane potential
- AM:
-
adrenomedullin
- AMPK:
-
AMP-activated protein kinase
- AS-ODNs:
-
antisense oligodeoxynucleotides (2DG, 2-deoxyglucose
- BFPP:
-
frontoparietal polymicrogyria
- BH:
-
Bcl-2 homology domain
- BA:
-
betulinic acid
- CT:
-
computed tomography
- Cyt c:
-
cytochrome c
- DLC:
-
delocalized lipophilic cations
- DcR1:
-
decoy TRAIL receptor 1
- DR:
-
death receptors
- ECM:
-
extracellular matrix
- ER:
-
endoplasmic reticulum
- EGCG:
-
(-)-epigallocatechin gallate
- GRP78:
-
glucose-regulated protein 78
- HIF-1α:
-
hypoxia-induced factor-1α
- HK II:
-
hexokinase II
- HSV-1:
-
herpes simplex virus 1
- HTS:
-
high throughput screening
- IAPs:
-
inhibitors of apoptosis proteins
- ILK:
-
integrin-linked kinase
- IM:
-
mitochondrial inner membrane
- IMS:
-
mitochondrial intermembrane space
- INF:
-
interferon
- LOF:
-
loss-of-function
- MMP:
-
mitochondrial membrane permeabilization
- NDV:
-
Newcastle disease virus
- OM:
-
mitochondrial outer membrane
- OXPHOS:
-
oxidative phosphorylation
- PET:
-
positron emission tomography
- PK:
-
protein kinase
- PLK:
-
polo-like kinase
- PTPC:
-
mitochondrial permeability transition pore complex
- RGD:
-
arginine-glycine-aspartic acid
- ROS:
-
reactive oxygen species
- TNF-α:
-
tumor necrosis factor-α
- TRAIL:
-
TNF-related apoptosis-inducing ligand
- TRAIL-R1:
-
TNF-related apoptosis-inducing ligand receptor 1
- UPR:
-
unfolded protein response
References
Fesik SW . Promoting apoptosis as a strategy for cancer drug discovery. Nat Rev Cancer 2005; 5: 876–885.
Grunhagen DJ, de Wilt JH, ten Hagen TL, Eggermont AM . Technology insight: utility of TNF-alpha-based isolated limb perfusion to avoid amputation of irresectable tumors of the extremities. Nat Clin Pract Oncol 2006; 3: 94–103.
Carlo-Stella C, Lavazza C, Locatelli A, Vigano L, Gianni AM, Gianni L . Targeting TRAIL agonistic receptors for cancer therapy. Clin Cancer Res 2007; 13: 2313–2317.
Huang Y, Sheikh MS . TRAIL death receptors and cancer therapeutics. Toxicol Appl Pharmacol 2007; 224: 284–289.
Marini P . Drug evaluation: lexatumumab, an intravenous human agonistic mAb targeting TRAIL receptor 2. Curr Opin Mol Ther 2006; 8: 539–546.
Camidge DR . The potential of death receptor 4- and 5-directed therapies in the treatment of lung cancer. Clin Lung Cancer 2007; 8: 413–419.
Li QX, Tan P, Ke N, Wong-Staal F . Ribozyme technology for cancer gene target identification and validation. Adv Cancer Res 2007; 96: 103–143.
Liu G, Wong-Staal F, Li QX . Recent development of RNAi in drug target discovery and validation. Drug Discov Today: Technologies 2006; 3: 337–341.
Kroemer G, Galluzzi L, Brenner C . Mitochondrial membrane permeabilization in cell death. Physiol Rev 2007; 87: 99–163.
Fulda S, Scaffidi C, Susin SA, Krammer PH, Kroemer G, Peter ME et al. Activation of mitochondria and release of mitochondrial apoptogenic factors by betulinic acid. J Biol Chem 1998; 273: 33942–33948.
Galluzzi L, Larochette N, Zamzami N, Kroemer G . Mitochondria as therapeutic targets for cancer chemotherapy. Oncogene 2006; 25: 4812–4830.
Oudard S, Carpentier A, Banu E, Fauchon F, Celerier D, Poupon MF et al. Phase II study of lonidamine and diazepam in the treatment of recurrent glioblastoma multiforme. J Neurooncol 2003; 63: 81–86.
Summerhayes IC, Lampidis TJ, Bernal SD, Nadakavukaren JJ, Nadakavukaren KK, Shepherd EL et al. Unusual retention of rhodamine 123 by mitochondria in muscle and carcinoma cells. Proc Natl Acad Sci USA 1982; 79: 5292–5296.
Davis S, Weiss MJ, Wong JR, Lampidis TJ, Chen LB . Mitochondrial and plasma membrane potentials cause unusual accumulation and retention of rhodamine 123 by human breast adenocarcinoma-derived MCF-7 cells. J Biol Chem 1985; 260: 13844–13850.
Nadakavukaren KK, Nadakavukaren JJ, Chen LB . Increased rhodamine 123 uptake by carcinoma cells. Cancer Res 1985; 45 (12 Part 1): 6093–6099.
Huang SG . Development of a high throughput screening assay for mitochondrial membrane potential in living cells. J Biomol Screen 2002; 7: 383–389.
Wong A, Cortopassi GA . High-throughput measurement of mitochondrial membrane potential in a neural cell line using a fluorescence plate reader. Biochem Biophys Res Commun 2002; 298: 750–754.
Fantin VR, Berardi MJ, Scorrano L, Korsmeyer SJ, Leder P . A novel mitochondriotoxic small molecule that selectively inhibits tumor cell growth. Cancer Cell 2002; 2: 29–42.
Shoemaker AR, Oleksijew A, Bauch J, Belli BA, Borre T, Bruncko M et al. A small-molecule inhibitor of Bcl-XL potentiates the activity of cytotoxic drugs in vitro and in vivo. Cancer Res 2006; 66: 8731–8739.
Hunter AM, LaCasse EC, Korneluk RG . The inhibitors of apoptosis (IAPs) as cancer targets. Apoptosis 2007; 12: 1543–1568.
Olie RA, Simoes-Wust AP, Baumann B, Leech SH, Fabbro D, Stahel RA et al. A novel antisense oligonucleotide targeting survivin expression induces apoptosis and sensitizes lung cancer cells to chemotherapy. Cancer Res 2000; 60: 2805–2809.
Marx J . Oncology. Recruiting the cell's own guardian for cancer therapy. Science (New York, NY) 2007; 315: 1211–1213.
Bykov VJ, Issaeva N, Shilov A, Hultcrantz M, Pugacheva E, Chumakov P et al. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat Med 2002; 8: 282–288.
Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science (New York, NY) 2004; 303: 844–848.
Gore SD, Jones C, Kirkpatrick P . Decitabine. Nat Rev Drug Discov 2006; 5: 891–892.
Tamm I, Wagner M, Schmelz K . Decitabine activates specific caspases downstream of p73 in myeloid leukemia. Ann Hematol 2005; 84 (Suppl 13): 47–53.
Reu FJ, Bae SI, Cherkassky L, Leaman DW, Lindner D, Beaulieu N et al. Overcoming resistance to interferon-induced apoptosis of renal carcinoma and melanoma cells by DNA demethylation. J Clin Oncol 2006; 24: 3771–3779.
Cai SX, Drewe J, Kasibhatla S . A chemical genetics approach for the discovery of apoptosis inducers: from phenotypic cell based HTS assay and structure-activity relationship studies, to identification of potential anticancer agents and molecular targets. Curr Med Chem 2006; 13: 2627–2644.
Mehlen P, Puisieux A . Metastasis: a question of life or death. Nat Rev Cancer 2006; 6: 449–458.
Bewick MA, Lafrenie RM . Adhesion dependent signalling in the tumour microenvironment: the future of drug targetting. Curr Pharm Des 2006; 12: 2833–2848.
Hannigan G, Troussard AA, Dedhar S . Integrin-linked kinase: a cancer therapeutic target unique among its ILK. Nat Rev Cancer 2005; 5: 51–63.
Legate KR, Montanez E, Kudlacek O, Fassler R . ILK, PINCH and parvin: the tIPP of integrin signalling. Nat Rev Mol Cell Biol 2006; 7: 20–31.
Cavallaro U . N-cadherin as an invasion promoter: a novel target for antitumor therapy? Curr Opin Investig Drugs 2004; 5: 1274–1278.
Ke N, Albers A, Claassen G, Yu DH, Chatterton JE, Hu X et al. One-week 96-well soft agar growth assay for cancer target validation. Biotechniques 2004; 36: 826–828, 830, 832–823.
Claassen G, Ke N, Yu D, Chatterton JE, Hu X, Albers A et al. Comprehensive assessment of cell growth properties using an integrated and nonsubjective approach. Preclinica 2004; 2: 435–439.
Hynes RO . Cell adhesion: old and new questions. Trends Cell Biol 1999; 9: M33–M37.
Ke N, Sundaram R, Liu G, Chionis J, Fan W, Rogers C et al. Orphan G protein-coupled receptor GPR56 plays a role in cell transformation and tumorigenesis involving the cell adhesion pathway. Mol Cancer Ther 2007; 6: 1840–1850.
Desoize B, Bouche O, Berthiot G, Coninx P . Puzzles in the clinical pharmacokinetics of fluorouracil. Anticancer Res 1998; 18: 4607–4610.
Esumi H, Lu J, Kurashima Y, Hanaoka T . Antitumor activity of pyrvinium pamoate, 6-(dimethylamino)-2-[2-(2,5-dimethyl-1-phenyl-1H-pyrrol-3-yl)ethenyl]-1-me thyl-quinolinium pamoate salt, showing preferential cytotoxicity during glucose starvation. Cancer Sci 2004; 95: 685–690.
Ke N, Claassen G, Yu DH, Albers A, Fan W, Tan P et al. Nuclear hormone receptor NR4A2 is involved in cell transformation and apoptosis. Cancer Res 2004; 64: 8208–8212.
Boylan MO, Athanassiou M, Houle B, Wang Y, Zarbl H . Activation of tumor suppressor genes in nontumorigenic revertants of the HeLa cervical carcinoma cell line. Cell Growth Differ 1996; 7: 725–735.
Yu DH, Fan W, Liu G, Nguy V, Chatterton JE, Long S et al. PHTS, a novel putative tumor suppressor, is involved in the transformation reversion of HeLaHF cells independently of the p53 pathway. Exp Cell Res 2006; 312: 865–876.
Bachelder RE, Ribick MJ, Marchetti A, Falcioni R, Soddu S, Davis KR et al. p53 inhibits alpha 6 beta 4 integrin survival signaling by promoting the caspase 3-dependent cleavage of AKT/PKB. J Cell Biol 1999; 147: 1063–1072.
Grossmann J . Molecular mechanisms of ‘detachment-induced apoptosis—Anoikis’. Apoptosis 2002; 7: 247–260.
Yu DH, Chatterton JE, Bliesath JR, Sundaram R, Ke N, Nguy V et al. A 96-well surrogate survival assay coupled with a special short interfering RNA vector for assessing cancer gene targets with enhanced signal/noise ratio and its utility in HTS for cancer therapeutic targets. Assay Drug Dev Technol 2005; 3: 401–411.
Li QX, Ke N, Sundaram R, Wong-Staal F . NR4A1, 2, 3 – an orphan nuclear hormone receptor family involved in cell apoptosis and carcinogenesis. Histol Histopathol 2006; 21: 533–540.
Suzuki S, Suzuki N, Mirtsos C, Horacek T, Lye E, Noh SK et al. Nur77 as a survival factor in tumor necrosis factor signaling. Proc Natl Acad Sci USA 2003; 100: 8276–8280.
Hsu HC, Zhou T, Mountz JD . Nur77 family of nuclear hormone receptors. Curr Drug Targets 2004; 3: 413–423.
Mullican SE, Zhang S, Konopleva M, Ruvolo V, Andreeff M, Milbrandt J et al. Abrogation of nuclear receptors Nr4a3 and Nr4a1 leads to development of acute myeloid leukemia. Nat Med 2007; 13: 730–735.
Chintharlapalli S, Burghardt R, Papineni S, Ramaiah S, Yoon K, Safe S . Activation of Nur77 by selected 1,1-Bis(3′-indolyl)-1-(p-substituted phenyl)methanes induces apoptosis through nuclear pathways. J Biol Chem 2005; 280: 24903–24914.
Ke N, Sundaram R, Liu G, Chionis J, Fan W, Rogers C et al. Orphan G-protein coupled receptor GPR56 plays a role in cell transformation and tumorigenesis involving the cell adhesion pathway. Mol Cancer Ther 2007; 6: 1840–1850.
Piao X, Hill RS, Bodell A, Chang BS, Basel-Vanagaite L, Straussberg R et al. G protein-coupled receptor-dependent development of human frontal cortex. Science (New York, NY) 2004; 303: 2033–2036.
Jin Z, Tietjen I, Bu L, Liu-Yesucevitz L, Gaur SK, Walsh CA et al. Disease-associated mutations affect GPR56 protein trafficking and cell surface expression. Hum Mol Genet 2007; 16: 1972–1985.
Ke N, Ma H, Diedrich G, Chionis J, Liu G, Yu DH et al. Biochemical characterization of genetic mutations of GPR56 in patients with bilateral frontoparietal polymicrogyria (BFPP). Biochem Biophys Res Commun 2008; 366: 314–320.
Shashidhar S, Lorente G, Nagavarapu U, Nelson A, Kuo J, Cummins J et al. GPR56 is a GPCR that is overexpressed in gliomas and functions in tumor cell adhesion. Oncogene 2005; 24: 1673–1682.
Xu L, Begum S, Hearn JD, Hynes RO . GPR56, an atypical G protein-coupled receptor, binds tissue transglutaminase, TG2, and inhibits melanoma tumor growth and metastasis. Proc Natl Acad Sci USA 2006; 103: 9023–9028.
Li QX, Robbins JM, Welch PJ, Wong-Staal F, Barber JR . A novel functional genomics approach identifies mTERT as a suppressor of fibroblast transformation. Nucleic Acids Res 2000; 28: 2605–2612.
Welch PJ, Marcusson EG, Li QX, Beger C, Kruger M, Zhou C et al. Identification and validation of a gene involved in anchorage-independent cell growth control using a library of randomized hairpin ribozymes. Genomics 2000; 66: 274–283.
Yang JP, Fan W, Rogers C, Chatterton JE, Bliesath J, Liu G et al. A novel RNAi library based on partially randomized consensus sequences of nuclear receptors: identifying the receptors involved in amyloid beta degradation. Genomics 2006; 88: 282–292.
Silva JM, Li MZ, Chang K, Ge W, Golding MC, Rickles RJ et al. Second-generation shRNA libraries covering the mouse and human genomes. Nat Genet 2005; 37: 1281–1288.
Lopez-Rios F, Sanchez-Arago M, Garcia-Garcia E, Ortega AD, Berrendero JR, Pozo-Rodriguez F et al. Loss of the mitochondrial bioenergetic capacity underlies the glucose avidity of carcinomas. Cancer Res 2007; 67: 9013–9017.
Aft RL, Zhang FW, Gius D . Evaluation of 2-deoxy-D-glucose as a chemotherapeutic agent: mechanism of cell death. Br J Cancer 2002; 87: 805–812.
Maschek G, Savaraj N, Priebe W, Braunschweiger P, Hamilton K, Tidmarsh GF et al. 2-deoxy-D-glucose increases the efficacy of adriamycin and paclitaxel in human osteosarcoma and non-small cell lung cancers in vivo. Cancer Res 2004; 64: 31–34.
Mohanti BK, Rath GK, Anantha N, Kannan V, Das BS, Chandramouli BA et al. Improving cancer radiotherapy with 2-deoxy-D-glucose: phase I/II clinical trials on human cerebral gliomas. Int J Radiat Oncol Biol Phys 1996; 35: 103–111.
Geschwind JF, Ko YH, Torbenson MS, Magee C, Pedersen PL . Novel therapy for liver cancer: direct intraarterial injection of a potent inhibitor of ATP production. Cancer Res 2002; 62: 3909–3913.
Andringa KK, Coleman MC, Aykin-Burns N, Hitchler MJ, Walsh SA, Domann FE et al. Inhibition of glutamate cysteine ligase activity sensitizes human breast cancer cells to the toxicity of 2-deoxy-D-glucose. Cancer Res 2006; 66: 1605–1610.
Barnes K, McIntosh E, Whetton AD, Daley GQ, Bentley J, Baldwin SA . Chronic myeloid leukaemia: an investigation into the role of Bcr-Abl-induced abnormalities in glucose transport regulation. Oncogene 2005; 24: 3257–3267.
Gottschalk S, Anderson N, Hainz C, Eckhardt SG, Serkova NJ . Imatinib (STI571)-mediated changes in glucose metabolism in human leukemia BCR-ABL-positive cells. Clin Cancer Res 2004; 10: 6661–6668.
Tada-Oikawa S, Hiraku Y, Kawanishi M, Kawanishi S . Mechanism for generation of hydrogen peroxide and change of mitochondrial membrane potential during rotenone-induced apoptosis. Life Sci 2003; 73: 3277–3288.
Bremer E, van Dam G, Kroesen BJ, de Leij L, Helfrich W . Targeted induction of apoptosis for cancer therapy: current progress and prospects. Trends Mol Med 2006; 12: 382–393.
Luo Z, Saha AK, Xiang X, Ruderman NB . AMPK, the metabolic syndrome and cancer. Trends Pharmacol Sci 2005; 26: 69–76.
Laderoute KR, Amin K, Calaoagan JM, Knapp M, Le T, Orduna J et al. 5′-AMP-activated protein kinase (AMPK) is induced by low-oxygen and glucose deprivation conditions found in solid-tumor microenvironments. Mol Cell Biol 2006; 26: 5336–5347.
Shaw RJ . Glucose metabolism and cancer. Curr Opin Cell Biol 2006; 18: 598–608.
Corradetti MN, Guan KL . Upstream of the mammalian target of rapamycin: do all roads pass through mTOR? Oncogene 2006; 25: 6347–6360.
McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ . Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol 2001; 21: 1249–1259.
Lievremont JP, Rizzuto R, Hendershot L, Meldolesi J . BiP, a major chaperone protein of the endoplasmic reticulum lumen, plays a direct and important role in the storage of the rapidly exchanging pool of Ca2+. J Biol Chem 1997; 272: 30873–30879.
Xu W, Liu L, Charles IG, Moncada S . Nitric oxide induces coupling of mitochondrial signalling with the endoplasmic reticulum stress response. Nat Cell Biol 2004; 6: 1129–1134.
Fernandez PM, Tabbara SO, Jacobs LK, Manning FC, Tsangaris TN, Schwartz AM et al. Overexpression of the glucose-regulated stress gene GRP78 in malignant but not benign human breast lesions. Breast Cancer Res Treat 2000; 59: 15–26.
Gazit G, Lu J, Lee AS . De-regulation of GRP stress protein expression in human breast cancer cell lines. Breast Cancer Res Treat 1999; 54: 135–146.
Song MS, Park YK, Lee JH, Park K . Induction of glucose-regulated protein 78 by chronic hypoxia in human gastric tumor cells through a protein kinase C-epsilon/ERK/AP-1 signaling cascade. Cancer Res 2001; 61: 8322–8330.
Koomagi R, Mattern J, Volm M . Glucose-related protein (GRP78) and its relationship to the drug-resistance proteins P170, GST-pi, LRP56 and angiogenesis in non-small cell lung carcinomas. Anticancer Res 1999; 19: 4333–4336.
Shuda M, Kondoh N, Imazeki N, Tanaka K, Okada T, Mori K et al. Activation of the ATF6, XBP1 and grp78 genes in human hepatocellular carcinoma: a possible involvement of the ER stress pathway in hepatocarcinogenesis. J Hepatol 2003; 38: 605–614.
Arap MA, Lahdenranta J, Mintz PJ, Hajitou A, Sarkis AS, Arap W et al. Cell surface expression of the stress response chaperone GRP78 enables tumor targeting by circulating ligands. Cancer Cell 2004; 6: 275–284.
Dong D, Ko B, Baumeister P, Swenson S, Costa F, Markland F et al. Vascular targeting and antiangiogenesis agents induce drug resistance effector GRP78 within the tumor microenvironment. Cancer Res 2005; 65: 5785–5791.
Gatenby RA . The potential role of transformation-induced metabolic changes in tumor-host interaction. Cancer Res 1995; 55: 4151–4156.
Jamora C, Dennert G, Lee AS . Inhibition of tumor progression by suppression of stress protein GRP78/BiP induction in fibrosarcoma B/C10ME. Proc Natl Acad Sci USA 1996; 93: 7690–7694.
Teicher BA . Hypoxia and drug resistance. Cancer Metastasis Rev 1994; 13: 139–168.
Mintz PJ, Kim J, Do KA, Wang X, Zinner RG, Cristofanilli M et al. Fingerprinting the circulating repertoire of antibodies from cancer patients. Nat Biotechnol 2003; 21: 57–63.
Ramsay RG, Ciznadija D, Mantamadiotis T, Anderson R, Pearson R . Expression of stress response protein glucose regulated protein-78 mediated by c-Myb. Int J Biochem Cell Biol 2005; 37: 1254–1268.
Misra UK, Deedwania R, Pizzo SV . Binding of activated alpha2-macroglobulin to its cell surface receptor GRP78 in 1-LN prostate cancer cells regulates PAK-2-dependent activation of LIMK. J Biol Chem 2005; 280: 26278–26286.
Marjon PL, Bobrovnikova-Marjon EV, Abcouwer SF . Expression of the pro-angiogenic factors vascular endothelial growth factor and interleukin-8/CXCL8 by human breast carcinomas is responsive to nutrient deprivation and endoplasmic reticulum stress. Mol Cancer 2004; 3: 4.
Fujimoto T, Yoshimatsu K, Watanabe K, Yokomizo H, Otani T, Matsumoto A et al. Overexpression of human X-box binding protein 1 (XBP-1) in colorectal adenomas and adenocarcinomas. Anticancer Res 2007; 27: 127–131.
Feldman DE, Chauhan V, Koong AC . The unfolded protein response: a novel component of the hypoxic stress response in tumors. Mol Cancer Res 2005; 3: 597–605.
Rao RV, Peel A, Logvinova A, del Rio G, Hermel E, Yokota T et al. Coupling endoplasmic reticulum stress to the cell death program: role of the ER chaperone GRP78. FEBS Lett 2002; 514: 122–128.
Lee E, Nichols P, Spicer D, Groshen S, Yu MC, Lee AS . GRP78 as a Novel predictor of responsiveness to chemotherapy in breast cancer. Cancer Res 2006; 66: 7849–7853.
Ermakova SP, Kang BS, Choi BY, Choi HS, Schuster TF, Ma WY et al. (-)-Epigallocatechin gallate overcomes resistance to etoposide-induced cell death by targeting the molecular chaperone glucose-regulated protein 78. Cancer Res 2006; 66: 9260–9269.
Park HR, Tomida A, Sato S, Tsukumo Y, Yun J, Yamori T et al. Effect on tumor cells of blocking survival response to glucose deprivation. J Natl Cancer Inst 2004; 96: 1300–1310.
Davidson DJ, Haskell C, Majest S, Kherzai A, Egan DA, Walter KA et al. Kringle 5 of human plasminogen induces apoptosis of endothelial and tumor cells through surface-expressed glucose-regulated protein 78. Cancer Res 2005; 65: 4663–4672.
Koyama AH, Fukumori T, Fujita M, Irie H, Adachi A . Physiological significance of apoptosis in animal virus infection. Microbes Infect 2000; 2: 1111–1117.
Parato KA, Senger D, Forsyth PA, Bell JC . Recent progress in the battle between oncolytic viruses and tumours. Nat Rev Cancer 2005; 5: 965–976.
Varghese S, Rabkin SD . Oncolytic herpes simplex virus vectors for cancer virotherapy. Cancer Gene Ther 2002; 9: 967–978.
Goodkin ML, Morton ER, Blaho JA . Herpes simplex virus infection and apoptosis. Int Rev Immunol 2004; 23: 141–172.
Fu X-P, Tao L, Zhang X . An HSV-2-based oncolytic virus deleted in the PK domain of the ICP10 gene is a potent inducer of apoptotic death in tumor cells. Gene Therapy 2007; 14: 1218–1225.
Fu X, Tao L, Cai R, Prigge J, Zhang X . A mutant type 2 herpes simplex virus deleted for the protein kinase domain of the ICP10 gene is a potent oncolytic virus. Mol Ther 2006; 13: 882–890.
Martuza RL, Malick A, Markert JM, Ruffner KL, Coen DM . Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science (New York, NY) 1991; 252: 854–856.
Mineta T, Rabkin SD, Yazaki T, Hunter WD, Martuza RL . Attenuated multi-mutated herpes simplex virus-1 for the treatment of malignant gliomas. Nat Med 1995; 1: 938–943.
Stanziale SF, Petrowsky H, Adusumilli PS, Ben-Porat L, Gonen M, Fong Y . Infection with oncolytic herpes simplex virus-1 induces apoptosis in neighboring human cancer cells: a potential target to increase anticancer activity. Clin Cancer Res 2004; 10: 3225–3232.
Elankumaran S, Rockemann D, Samal SK . Newcastle disease virus exerts oncolysis by both intrinsic and extrinsic caspase-dependent pathways of cell death. J Virol 2006; 80: 7522–7534.
Kominsky DJ, Bickel RJ, Tyler KL . Reovirus-induced apoptosis requires both death receptor- and mitochondrial-mediated caspase-dependent pathways of cell death. Cell Death Differ 2002; 9: 926–933.
Acknowledgements
We thank Dr Helmut Zarbl for kindly providing HeLaHF cells.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by RA Knight
Rights and permissions
About this article
Cite this article
Li, QX., Yu, D., Liu, G. et al. Selective anticancer strategies via intervention of the death pathways relevant to cell transformation. Cell Death Differ 15, 1197–1210 (2008). https://doi.org/10.1038/cdd.2008.48
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2008.48
Keywords
This article is cited by
-
Antiproliferative and Apoptosis-Inducing Activities of Thymoquinone in Lymphoblastic Leukemia Cell Line
Indian Journal of Hematology and Blood Transfusion (2017)
-
Oxidative phosphorylation-dependent regulation of cancer cell apoptosis in response to anticancer agents
Cell Death & Disease (2015)
-
Suppression of FOXO1 is responsible for a growth regulatory repressive transcriptional sub-signature of EWS-FLI1 in Ewing sarcoma
Oncogene (2014)
-
Necroptosis: Biochemical, Physiological and Pathological Aspects
Pathology & Oncology Research (2011)
-
Differential gene expression and clonal selection during cellular transformation induced by adhesion deprivation
BMC Cell Biology (2010)
