
Abstract
Background:
The direct comparison of CA19.9, circulating tumour cells (CTCs) and circulating tumour DNA (ctDNA) using endoscopic ultrasound-guided fine needle aspiration (EUS-FNA) has never been performed for the diagnosis of solid pancreatic tumours (SPTs).
Methods:
We included 68 patients with a SPT referred for EUS-FNA. CTCs were analysed using size-based platform and ctDNA using digital PCR. The sensitivity, specificity, negative and positive predictive values were evaluated for each marker and their combination.
Results:
SPTs corresponded to 58 malignant tumours (52 pancreatic adenocarcinoma (PA) and 6 others) and 10 benign lesions. The sensitivity and specificity for PA diagnosis were 73% and 88% for EUS-FNA, 67% and 80% for CTC, 65% and 75% for ctDNA and 79% and 93% for CA19.9, respectively. The positivity of at least 2 markers was associated with a sensitivity and specificity of 78% and 91%, respectively. CtDNA was the only marker associated with overall survival (median 5.2 months for ctDNA+ vs 11.0 months for ctDNA−, P=0.01).
Conclusions:
CA19.9 alone and in combination with ctDNA and/or CTC analysis may represent an efficient method for diagnosing PA in patients with SPTs. Further studies including a larger cohort of patients with both malignant and benign lesions will be necessary to confirm these promising results.
Similar content being viewed by others


Main
Pancreatic adenocarcinoma (PA) is the fourth leading cause of cancer-related death worldwide, with a poor 5-year survival rate of <10% across all stages. PA represents 90% of pancreatic tumours; the other tumours are benign lesions with a pseudotumoral form of chronic pancreatitis or malignant forms such as an endocrine tumour, haematological malignancy or metastasis (Kamisawa et al, 2016). At presentation, ∼80% of patients with PA have a locally advanced or metastatic disease, and histological confirmation is usually required before palliative chemotherapy and/or radiotherapy. In this context, endoscopic ultrasound-guided fine needle aspiration (EUS-FNA) is considered a standard for PA confirmation in patients presenting an isolated pancreatic tumour (ASGE Standards of Practice Committee et al, 2016), with a sensitivity and specificity ranging from 78 to 95% and from 75 to 100%, respectively (Yoshinaga et al, 2011). The procedure needs to be repeated in ∼20–30% of cases, leading to a potential delay for diagnostic confirmation (Tadic et al, 2008).
To date, the carbohydrate antigen CA19.9 (CA19.9), which has been approved by the FDA for PA management, is the only marker used in daily practice. Its clinical interest has been intensively studied, and it has a sensitivity of 70–81% for PA diagnosis (Chan et al, 2014; Su et al, 2015). The well-known limitation of CA19.9 is its low specificity due to the presence of elevated levels in non-malignant situations, such as cholestasis or diabetes mellitus. In this context, a liquid biopsy based on circulating tumour DNA (ctDNA) and circulating tumour cells (CTCs), which result from metastatic or primary tumour release, are considered very promising to optimise the diagnosis and decision making in PA patients. CTCs are rare cells that disseminate into the bloodstream, and their detection is usually performed by a combination of enrichment and detection methods. Cell-free DNA (cfDNA) corresponds to all DNA fragments resulting from both normal and tumour cells, whereas ctDNA harbors specific genetic alterations from tumour cells (Diaz and Bardelli, 2014). The detection of ctDNA requires ultrasensitive technologies, including next-generation sequencing-based or digital PCR-based platforms with a sensitivity up to 0.01% of the allelic frequency and a detection rate ranging from 26 to 90% in PA (Bettegowda et al, 2014; Earl et al, 2015; Kinugasa et al, 2015; Sausen et al, 2015; Takai et al, 2015; Zill et al, 2015; Brychta et al, 2016).
To date, the usefulness of CTC and ctDNA detection has not been clearly established in PA patients. We previously reported that CTC detection using the Screencell method had a sensitivity and specificity of 55% and 100%, respectively, for PA diagnosis (Iwanicki-Caron et al, 2013). Similar to colorectal cancer, the ctDNA level seems to be associated with the disease stage and may have potential prognostic impacts in patients presenting with a PA (Earl et al, 2015; Kinugasa et al, 2015; Sausen et al, 2015; Takai et al, 2015).
To the best of our knowledge, the diagnostic value of CA19.9 compared with CTC and circulating DNA, as well as their combination, has never been evaluated in PA patients. We conducted a study to investigate their relevance alone or in combination in patients who underwent EUS-FNA for a pancreatic solid tumour.
Materials and methods
Patients and sample collection
Patients undergoing EUS-FNA for the exploration of a solid pancreatic tumour in Rouen University Hospital from January 2011 to March 2014 were included after their informed consent. No patients had received treatment before the EUS-FNA. PA was classified as localised disease (LD), locally advanced disease (LAD) or metastatic disease (MD) based on a CT-scan. For each patient, blood samples were collected into a single EDTA tube (4 ml) for CTC analysis and one heparinised tube (4 ml) for CA19.9 and circulating DNA (cDNA) analysis immediately before the EUS-FNA procedure. The CA19.9, CTC and cDNA analyses were centralised, and the study was approved by the institutional review board (Northwest I).
EUS-FNA technique
The EUS-FNA procedures were performed under general anesthesia by three experimented endoscopists. The echoendoscopic examination was performed with radial (GF-UE160-AL5, Olympus, Tokyo, Japan) and/or linear (GF-UC140P-AL5, Olympus, Tokyo, Japan) echoendoscopes, and cytological samples were obtained using three back-and-forth passages with 22 G needles. The samples was placed in Hank's solution, fixed in AFA (absolute alcohol: 50%, 40% formaldehyde: 20%, acetic acid: 5%, and distilled water: 25%) and embedded in paraffin. The slides were stained with hematoxylin-eosin-safran and analysed by a pathologist who classified the samples as follows: positive for malignancy, suspicious for malignancy, negative for malignancy or benign lesion and non-contributive. In cases of non-contributive FNA, the histological confirmation was obtained using a second FNA, biopsy or a surgical procedure according to the clinical context.
Determination of the KRAS mutational status in tumour tissue
DNA for the KRAS mutational status determination was based on the most adequate genetic material when available (surgical specimen>biopsy>paraffin-embedded section of the cell blocks obtained from the EUS-FNA). After extraction, genotyping of KRAS mutations within exon 2 (codons 12 and 13) was performed using the SNaPshot multiplex assay, which required at least 5% tumour cells, as previously described (Lamy et al, 2011).
CA 19.9 analysis
CA19.9 was measured on a Kryptor PLC (B.R.A.H.M.S., Hennigsdorf, Germany) by the immunological method in the homogeneous phase. The upper limit of the normal value was 35 UI ml−1.
Circulating total DNA
cDNA analysis was performed after centrifuging the peripheral blood samples at 2700 g for 20 min at room temperature and then at 16 000 g for 10 min at 4 °C. The second step was performed before extraction and after the heparinised plasma sample previously collected for CA19.9 analysis was thawed. DNA was extracted from 0.35 to 1.7 ml of heparinised plasma using the QIamp Circulating Nucleic Acid kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Data regarding the influence of the initial plasma volume on cDNA analysis are shown in Supplementary Figures 1–4 and Supplementary Table 1). The sample was eluted in a final volume of 30 μl and stored at −20 °C. Quantification of the cfDNA was performed using a fluorometric method as previously described (Sefrioui et al, 2015a, 2015b). The concentration was expressed as ng ml−1 of plasma using the following equation: CP=CExVE/VP, where CE corresponds to the concentration of extracted cfDNA (ng μl−1), CP corresponds to the concentration of cfDNA in the plasma (ng ml−1), VE corresponds to the elution volume used for the cfDNA extraction (μl) and VP corresponds to the plasma volume used to extract the cfDNA (ml). Consequently, the cfDNA concentrations could be compared between the patients.
Circulating tumour DNA
The detection of ctDNA was based on KRAS mutations. First, preamplification was performed since this step was not associated with any qualitative and quantitative alterations (including mutant allelic fraction) in the dPCR results compared with those obtained from the original sample (Wang et al, 2015; Jackson et al, 2016) (Supplementary Figure 5). Thus, a preamplification using four ng of cfDNA was performed in a final volume of 25 μl consisting of 12.5 μl of 2X TaqMan genotyping master mix (Applied Biosystems, Foster City, CA, USA), 0.25 μl of the primer/probe mixture (Custom TaqMan SNP Genotyping Assays for c.35G>A detection, Life Technologies, Carlsbad, CA, USA) and 2 μl of heparinase (New England Biolabs, Ipswich, MA, USA). According to the results of previous runs showing an inhibitory effect of heparin on preamplification, heparinase was systematically added to inhibit the action of heparin in the PCR (Beutler et al, 1990). All runs were performed using the following thermocycling conditions: 10 min at 95 °C, followed by 15 cycles of 15 s at 95 °C and 4 min at 60 °C. The droplet-based dPCR (ddPCR) platform (Qx200 ddPCR system, Bio-Rad, Hercules, CA, USA) was used for ctDNA detection. A multiplex assay (Bio-Rad, Hercules, CA, USA) was used to screen the most common KRAS mutations found in PA (n=7: c.34G>A, c.34G>C, c.34G>T, c.35G>A, c.35G>C, c.35G>T and c.38G>A). A simplex assay (Life Technologies, Carlsbad, CA, USA) based on specific KRAS mutations was also performed to confirm the results of positive samples obtained using the multiplex assay. The ddPCR conditions and precisions concerning the limit of detection are described in Supplementary Materials. Two simplex assays (c.34G>A and c.34G>C) failed because no amplification could be achieved for DNA from heparinised plasma. The ddPCR analysis was performed blindly from clinical data.
Circulating tumour cell analysis
Blood samples were conserved at room temperature and processed within 3 h of collection. CTC isolation was performed using the size-based Screencell Cyto filtration device according to manufacturer’s instructions as previously reported (Iwanicki-Caron et al, 2013; Kulemann et al, 2015). Briefly, 4 ml of the ScreenCell FC filtration buffer was incubated for 8 min with 3 ml of blood to fix the target cells and lyse the red blood cells (RBCs). Then, the mixture was drawn through the filter by a vacuum tube. Following the filtration of the fixed cells, an additional 1.6 ml of 1 × phosphate-buffered saline was filtered to remove debris from the filter. Cell cytology was visualised with hematoxylin-eosin-safran staining. Enumeration and analysis of the structural characterisation of the CTCs was performed by an experienced cytopathologist blinded to the histological diagnosis. The cells were classified as ‘positive’ or ‘negative’ for CTC based on cytomorphological features. The cells were considered positive (i.e., circulating tumour cells) by the presence of at least four of the following criteria as previously described (Hofman et al, 2012): anisonucleosis (ratio>0.5), nuclei larger than the calibrated pore size of the membrane (8 μm), irregular nuclei, high nucleo-cytoplasmic ratio, and presence of three-dimensional sheets (Supplementary Figure 6). Cells were considered negative (i.e., circulating non-tumour cells) by the absence of at least four criteria.
Statistical analysis
The sensitivity, specificity, and negative and positive predictive values for the PA diagnosis were evaluated for EUS-FNA, which was considered the standard method, and for each marker. The performance of combinations, based on at least two positive markers among CA19.9, CTCs and ctDNA, was also analysed. A CA19.9 value above the normal limit and/or CTC⩾1 and/or detectable ctDNA based on the circulating multiplex KRAS analysis were the cut-offs used for the analysis of the combinations.
The prognostic impact was analysed for overall survival (OS) in PA patients. For the purpose of the study, the diagnosis of malignant tumours (i.e., PA or other malignant tumours) was defined either by histological evidence obtained by EUS-FNA and/or surgical specimens or by clinical outcomes with metastatic evolution and the CA19.9 serum level rather than 10-fold the normal value for PA. Benign lesions (i.e., chronic pancreatitis) were determined by follow-up of >3 years without any evidence of progression and EUS-FNA classification of the sample as indeterminate or negative in at least one procedure. The OS was calculated from the time of EUS-FNA to death from any cause. Patients were followed until April 30, 2016. The characteristics of patients and between predefined groups were expressed as percentages or means±s.d. for continuous variables and were compared using the Chi-square test, Fisher's exact test and Student’s T test as appropriate. Survival curves were calculated using the Kaplan–Meier method and compared with the log-rank test. The statistical analyses were calculated using StatView version 5.0 (SAS Institute Inc., Cary, NC, USA).
Results
Patient characteristics
A total of 68 patients presenting with a solid pancreatic tumour were included; their characteristics are listed in Table 1. The diagnosis corresponded to 58 (85%) malignant tumours (52 PA and 6 other malignant tumours) and 10 (15%) benign lesions (Table 1). For the 52 patients with PA, 13 had a LD, 17 had a LAD and the 22 remaining patients had a MD. Surgical resection of the tumour was performed in 9 patients, including 5 with PA, 3 with others malignant tumours (1 pancreatic neuroendocrine tumour (PanNET), 1 renal clear cell carcinoma metastasis and 1 fibrolamellar hepatocellular carcinoma with pancreas invasion) and 1 with a benign tumour corresponding to a pseudo-papillary tumour of the pancreas. The follow-up period was 25 months for all patients (April 2016) after the last patient was included (March 2014). At the time of the analysis, the median follow-up was 7.5 months (range 1–64), and 52 (76.5%) deaths were observed.
EUS-FNA results
A total of 86 EUS-FNA procedures were performed, including 1 procedure for 52 patients, 2 for 14 patients and 3 for 2 patients (mean=1.3±0.5). The diagnostic accuracy of the first EUS-FNA for PA was 76%, corresponding to a sensitivity and specificity of 73% and 88%, respectively (Table 2). The final diagnosis was based on EUS-FNA for 50 patients (first EUS-FNA, n=39; second or third EUS-FNA, n=11), a hepatic or surgical biopsy for 6 patients, surgical specimens for 9 patients and clinical outcomes with the CA19.9 level rather than 10-fold the normal value for the 3 remaining cases. There were two false positive results for PA, corresponding to one PanNET and one renal clear cell carcinoma metastasis on a surgical specimen.
CA19.9
CA 19.9 measurements were available for 63/68 (92.6%) patients, with a median value of 90 UI ml−1 (range 4–181 761) in all patients and 174 UI ml−1 (range 4–181 761) in PA. The mean CA19.9 concentration was not significantly different in patients with or without cholestasis (3087±9059 and 3587 ±12 935, respectively, P=0.92). The mean concentrations were 7435±28 102, 24±35, and 20±11 in PA, other malignant tumours and benign lesions, respectively (P=0.25). The difference between the mean and median values was due to the skewed distribution of the CA19.9 values, with 5 patients presenting CA19.9 values greater than 10 000 UI/ml (13 759; 16 490; 45 106; 64 095 and 181 761 UI ml−1) in PA group; conversely, the maximum CA19.9 in the other groups did not exceed 94 UI ml−1. The CA19.9 level was greater than the normal value in 79% (38/48) of the patients with PA malignant tumours, 17% (1/6) of the patients with non-PA malignant tumours and 0% (0/9) of the patients with benign lesions (P<0.001). The sensitivity and specificity of CA19.9 for the PA diagnosis were 79% and 93%, respectively (Table 2).
According to the liver test, the sensitivity and specificity were superior for PA diagnosis in patients without cholestasis (81% and 100%, respectively) vs those with cholestasis (74% and 67%, respectively) (Supplementary Tables 2 and 3). There was no correlation between the median CA19.9 (⩾174 UI/ml) and the baseline clinical parameters.
CTC detection
The analysis was informative for 64/68 samples (94%), with filtration difficulties for the 4 remaining cases. CTCs were detected (⩾1 CTC ml−1) in 36/64 samples (56% of the study population) compared with 33/49 (67%) patients with PA. The median CTC levels were 1 CTC ml−1(range 0–151) vs 4 CTC ml−1 (range 0–151) in these two groups, respectively. As shown in Table 2, the sensitivity and specificity for the PA diagnosis were 67% and 80%, respectively. There was a trend for increased CTC detection with tumour stage, with positive CTC detected in 16/28 (57%) patients with LD/LAD vs 17/21 (81%) patients with MD (P=0.12) (Supplementary Table 4). There was no correlation between the presence of CTC or the median CTC (⩾4 CTC ml−1) and the baseline clinical parameters. There were three false positive PA results, corresponding to one PanNET and two benign lesions.
Total and tumour circulating DNA analyses
Quantification of cfDNA was expressed as ng ml−1 of plasma and was successfully performed in 56 (82%) patients, with a median value of 57.6 ng ml−1 (range 12.9–925.3) in all patients and 59.5 ng ml−1 (range 12.9–925.3) in PA. The mean cfDNA concentrations were 99.2±137.2 ng ml−1 and 59.1±28.4 ng ml−1 in patients with malignant tumours (PA and others) and benign lesions, respectively (P=0.42). The mean cfDNA concentration was not significantly different according to the PA stage, with 73.8±45.6, 77.2±41.1 and 122.4±44 in the localised, locally advanced and metastatic diseases, respectively.
A KRAS circulating mutation was detected in 31/55 (56%) cases with available blood samples, corresponding to a ctDNA median value of 0.6% allelic frequency (range 0.17–41.1%) in all patients and 0.75% allelic frequency (range 0.33–41.1%) in PA. The sensitivity and specificity of ctDNA for the PA diagnosis were 65% and 75%, respectively (Table 2). There were 3 false positive PA results, corresponding to 1 PanNET and 2 benign lesions. There was a significant increase in ctDNA detection associated with tumour stage, with ctDNA present in 10/23 (43%) patients with LD/LAD vs 18/20 (90%) patients with MD (P=0.002) (Supplementary Table 4). Among the 27 PA patients with available paired plasma and tumour tissue samples, a KRAS mutation was found in 17/27 (63%) plasma samples. The concordance of the KRAS mutational status between the tissue and plasma was 19/27 (70.4%) (Table 3). For the three patients with a KRAS mutation in circulation and a wild-type KRAS status in the tumour tissue, re-examination of the blade used for determine the KRAS status in the tumour tissue showed low tumoural cellularity (i.e., <20%). For the 31 plasma samples in which a KRAS mutation was identified with the multiplex assay, 23 (74%) were positive using the simplex assays (Supplementary Table 5) with the following mutational profiles: c.34G>T (n=1), c.35G>A (n=13), c.35G>T (n=7), and c.38G>A (n=6). The multiplex and simplex assay results were significantly correlated (Figures 1 and 2). There was no correlation between the cfDNA median (⩾59.5 ng ml−1) and the baseline clinical parameters. However, the presence of ctDNA was significantly associated with a larger primary tumour size (83% for tumours ⩾30 mm vs 42% for tumours <30 mm, P=0.01).

Representation of circulating tumour DNA detection with ddPCR for two patients with pancreatic adenocarcinoma and a KRAS mutation (p.G12D for patient 47 and p.G12V for patient 63). The graphs in (A and C) illustrate the data obtained with multiplex assays, and the graphs in (B and D) illustrate the data obtained with simplex assays. The blue and green dots represent droplets containing amplified mutant and wild-type alleles, respectively. The red dots correspond to droplets containing both mutant and wild-type alleles. The gray dots correspond to wells containing no alleles (no amplification). % circulating tumour DNA represents the fraction of mutant allele/total allele (wild-type allele+mutant allele). Abbreviation: ctDNA, circulating tumour DNA.

Comparison of results obtained with the multiplex and simplex analyses. The graph illustrates the concordance of the allelic frequency (r2=0.995) between the 23 samples that tested positive in the multiplex and simplex analyses.
Marker combinations
The combined analysis of three markers was available in 51 (75%) patients, and 32 (63%) had at least two positive markers. The sensitivity and specificity for the PA diagnosis of the combinations were 78% and 91%, respectively (Table 2). The sensitivity was similar in patients with and without cholestasis (78% and 79%, respectively), whereas the specificity was superior in patients with cholestasis (100% vs 86%, respectively).
Circulating markers and outcomes
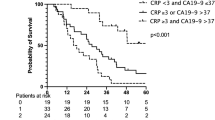
The median OS in patients with PA was 5.6 months (range 0.1–61.9). The median OS was 11.1 months in patients with LD compared with 7.4 months and 2.6 months in patients with LAD and MD, respectively (P=0.006). Patients with PA and detectable ctDNA had a significantly shorter OS compared with patients without detectable ctDNA (5.2 months vs 11 months, respectively, P=0.01) (Figure 3). A high ctDNA value (above vs below the median) in PA patients was also significantly associated with a poorer outcome, with a median OS of 3.9 months vs 7.8 months, respectively (P= 0.04) (Figure 3). The ctDNA analysis showed a decrease in the OS by quartile repartition (>75%, 50–75%, 25–50%, and <25%), with a median OS of 2.4 months, 6.1 months, 7.4 months and 11 months, respectively (P=0.0004). There was no correlation between the other markers and the outcome in patients with PA and other lesions (Figure 3).

Overall survival of patients with pancreatic adenocarcinoma according to circulating biomarkers at baseline. (A) Kaplan–Maier survival curve stratified by the presence (black line) or absence (red line) of CTCs. (B) Kaplan–Maier survival curve stratified by the CTC level above (black line) or below (red line) the median value (4 CTC ml−1). (C) Kaplan–Maier survival curve stratified by the presence (black line) or absence (red line) of ctDNA. (D) Kaplan–Maier survival curve stratified by the ctDNA level above (black line) or below (red line) the median allelic frequency value (0.75%). (E) Kaplan–Maier survival curve stratified by the cfDNA level above (black line) or below (red line) the median value (59.5 ng ml−1) expressed as ng ml−1 of plasma. (F) Kaplan–Maier survival curve stratified by the CA19.9 level above (black line) or below (red line) the median value (174 UI l−1).
Discussion
Our results suggest that CA19.9 alone and in combination with ctDNA and CTC analyses may represent a simple and efficient approach for PA diagnosis in patients presenting with SPTs with a sensitivity and specificity of their combination in the whole population of 78% and 91%, vs 73% and 88% for EUS-FNA alone, respectively. Moreover, considering the sample size for the subgroup analysis, CA19.9 performance decreased in cases of cholestasis, whereas the combination of markers remained relevant, despite the liver test. Moreover, we observed that a high ctDNA level at presentation may be associated with shorter survival in these patients. To the best of our knowledge, these markers have never been investigated together in patients undergoing an EUS-FNA for a solid pancreatic tumour. Because EUS-FNA may have some limitations, such as the need for general anesthesia, our results indicate that the analysis of these markers may provide relevant information for both diagnosis and prognosis.
KRAS mutations were logically used as targets for ctDNA detection because KRAS is the most commonly mutated gene in PA (90%), with a hotspot region (codons 12/13) covering 90% of KRAS mutations (Forbes et al, 2017). On the basis of a multiplex assay covering seven mutations in the hotspot region, we found a detection rate of 65% in PA patients, with an overall concordance of 70.4% in the subgroup with available paired tissue and blood samples. The ddPCR method developed in the present study showed a detection rate that was slightly superior to the rate that was recently reported (26–63%) (Earl et al, 2015; Kinugasa et al, 2015; Takai et al, 2015). In our work, a preamplification step was performed to improve mutant fragment DNA recovery (Jackson et al, 2016). Moreover, several clinical parameters, such as disease stage and prior treatment (Earl et al, 2015), may be associated with ctDNA detection (Bettegowda et al, 2014; Takai et al, 2015). In the present study, approximately 75% of the cases had advanced PA, although all patients were treatment naïve at the time of analysis. Finally, the release of different KRAS subclones into the plasma (Azuara et al, 2012; Visani et al, 2013; Kinugasa et al, 2015) due to tumour heterogeneity may also interfere with the ddPCR results, as indicated by our results in which duplicate or triplicate circulating mutations were detected in three cases. Taken together, these findings emphasised that ctDNA appeared to be an important marker in PA patients and that biological, clinical and preanalytical conditions are key factors involved in its detection. On the basis of its prognostic impact in our series and in other studies (Kinugasa et al, 2015; Sausen et al, 2015; Singh et al, 2015; Takai et al, 2015; Hadano et al, 2016), further prospective studies are needed to determine the most appropriate conditions for ctDNA detection as well as its clinical impact in PA patients.
We detected CTCs in 56% of PA patients, which was concordant with previous reports using size-based enrichment methods (Khoja et al, 2012; Kulemann et al, 2015; Poruk et al, 2016). The performance of CTC detection may be variable across techniques, with lower results obtained with immunological capture methods such as the Cellsearch technology(Kurihara et al, 2008; Khoja et al, 2012; Bidard et al, 2013), probably due to the small fraction of CTCs that express epithelial markers, such as EpCAM, in PA (Grover et al, 2014). As shown by studies based on sized-based enrichment methods, we did not observe differences in survival between patients with positive CTC vs negative CTC results. To date, the prognostic value of CTC in PA patients remains a subject of debate. Indeed, although some studies using immunological capture (Kurihara et al, 2008; Bidard et al, 2013) reported a decrease in survival in the presence of CTCs, a recent work suggested that a subset of CTCs (i.e., epithelial phenotype) as opposed to all CTCs had a poorer prognostic impact in PA patients (Poruk et al, 2016). To summarise, our results suggest that CTC detection may be useful for PA diagnosis, whereas their detection provides no relevant prognostic information.
We observed that CA19.9, which has been widely investigated in pancreatic illness (Winter et al, 2013), was also the most relevant marker for PA diagnosis, with a sensitivity of 79% and specificity of 93%. Two meta-analyses found similar values, with 79–81% sensitivity and 82–90% specificity (Ballehaninna and Chamberlain, 2012; Su et al, 2015). However, the usefulness of CA19.9 for PA diagnosis is still debated because of its low specificity. This value is elevated in several conditions, including chronic pancreatitis or cholestasis. An increase in CA19.9 was described in 15–20% of chronic pancreatitis without associated-cholestasis or PA (Nouts et al, 1998; Slesak et al, 2000). Normal values in the presence of PA have also been reported in 5–10% of the population due to the lack of Lewis antigen expression. Because 63% of the PA patients in our work presented concomitant cholestasis at the initial EUS-FNA, the interpretation of the CA19.9 results needs to be taken with caution. Indeed, diagnosis values of CA19.9 were particularly interesting in patients without cholestasis (sensitivity and specificity of 81% and 100%, respectively), whereas the performance decreased in patients with cholestasis (sensitivity and specificity of 74% and 67%, respectively) (Supplementary Tables 2 and 3). To overcome this limitation, we analysed the CTC and ctDNA in combination with CA19.9. The results showed that the presence of at least two markers was associated with a sensitivity and specificity of 78% and 91%, respectively. In patients with cholestasis, the combination improved the diagnosis accuracy compared with CA19.9 alone (79% vs 73%, respectively) (Supplementary Table 2). Interestingly, the positive predictive value of the combination was 97% for PA compared with 93% for EUS-FNA. In light of these results, we believe that these circulating makers may be useful for diagnosing PA and that further studies are needed to evaluate the relevance of their detection alone and as a complement to EUS-FNA. Indeed, we showed that the association of EUS-FNA with CA19.9, which was the most efficient biomarker in our series, was associated with an increase in sensitivity (100%) but a loss of specificity (80%). The additional cost of EUS-FNA remains exorbitant compared with the cost for the analysis of all 3 biomarkers (i.e., <$300). Moreover, circulating approach could be performed the same day of the physician’s visit.
The present study has some limitations. First, the patient cohort included few patients with benign lesions, which limits any definitive conclusions from these results, and additional larger studies are needed to confirm our results. The second limitation concerns the specificity of all three biomarkers, which have been reported to be associated with other solid tumours such as colon cancer. For example, increases in CA19.9 have been identified in gastric, colonic and esophageal adenocarcinoma at rates of 41%, 34% and 22%, respectively (Steinberg, 1990). However, we analysed these markers in patients presenting an SPT. Pancreatic metastases represent a very rare etiology among SPT and occur in <2% of all cases (Harewood and Wiersema, 2002; Varadarajulu et al, 2005; Uehara et al, 2011). Considering that majority of these SPTs originate from renal carcinoma (which are exceptionally associated with increased CA19.9 expression or KRAS mutation (DeWitt et al, 2005; Sweeney et al, 2009; Forbes et al, 2017), we think that these markers are specific in the PA diagnosis for patients with SPTs. Another limitation concerns the preanalytical conditions (including the variable initial plasma volume), which may have affected the results of the cDNA analysis. Because of this important issue, we performed subsequent analyses showing that the plasma volume used for cDNA extraction did not appear to significantly influence the cDNA (total and tumour) results (see Supplementary Figures 1–4). Thus, prospective trials will be necessary to clarify the optimal preanalytical and analytical conditions used for the ctDNA and CTC analyses in these settings. Finally, we observed an EUS-FNA sensitivity of 73% in our population. This result is slightly inferior to previously reported results (Eloubeidi et al, 2003a, 2003b; Varadarajulu et al, 2005) and may be due in part to our EUS-FNA process, which was based on 3–4 fine needle passages, whereas several works reported better results using 5–7 passages (LeBlanc et al, 2004).
In conclusion, our results indicate that CA19.9 in combination with CTC and ctDNA analyses represents an interesting alternative for PA diagnosis in patients with SPTs. Indeed, if individual analyses of CTC or ctDNA do not appear to be superior to EUS-FNA for diagnosing PA, a combination of markers seems to be a more promising approach with higher sensitivity and specificity than EUS-FNA alone. We also found that ctDNA may reflect the tumour burden in patients with solid pancreatic tumours. These results need to be confirmed, and a prospective study is currently ongoing in our center (NCT02072616) to confirm the diagnostic and prognostic values of CA19.9, CTC and ctDNA in patients with PA.
References
ASGE Standards of Practice Committee, Eloubeidi MA, Decker GA, Chandrasekhara V, Chathadi KV, Early DS, Evans JA, Fanelli RD, Fisher DA, Foley K, Hwang JH, Jue TL, Lightdale JR, Pasha SF, Saltzman JR, Sharaf R, Shergill AK, Cash BD, DeWitt JM ( 2016 ) The role of endoscopy in the evaluation and management of patients with solid pancreatic neoplasia . Gastrointest Endosc 83 : 17 – 28 .
Azuara D, Ginesta MM, Gausachs M, Rodriguez-Moranta F, Fabregat J, Busquets J, Pelaez N, Boadas J, Galter S, Moreno V, Costa J, de Oca J, Capellá G ( 2012 ) Nanofluidic digital PCR for KRAS mutation detection and quantification in gastrointestinal cancer . Clin Chem 58 : 1332 – 1341 .
Ballehaninna UK, Chamberlain RS ( 2012 ) The clinical utility of serum CA 19-9 in the diagnosis, prognosis and management of pancreatic adenocarcinoma: An evidence based appraisal . J Gastrointest Oncol 3 : 105 – 119 .
Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, Bartlett BR, Wang H, Luber B, Alani RM, Antonarakis ES, Azad NS, Bardelli A, Brem H, Cameron JL, Lee CC, Fecher LA, Gallia GL, Gibbs P, Le D, Giuntoli RL, Goggins M, Hogarty MD, Holdhoff M, Hong S-M, Jiao Y, Juhl HH, Kim JJ, Siravegna G, Laheru DA, Lauricella C, Lim M, Lipson EJ, Marie SKN, Netto GJ, Oliner KS, Olivi A, Olsson L, Riggins GJ, Sartore-Bianchi A, Schmidt K, Shih LE-M, Oba-Shinjo SM, Siena S, Theodorescu D, Tie J, Harkins TT, Veronese S, Wang T-L, Weingart JD, Wolfgang CL, Wood LD, Xing D, Hruban RH, Wu J, Allen PJ, Schmidt CM, Choti MA, Velculescu VE, Kinzler KW, Vogelstein B, Papadopoulos N, Diaz LA Jr ( 2014 ) Detection of circulating tumor DNA in early- and late-stage human malignancies . Sci Transl Med 6 : 224ra24 .
Beutler E, Gelbart T, Kuhl W ( 1990 ) Interference of heparin with the polymerase chain reaction . BioTechniques 9 : 166 .
Bidard FC, Huguet F, Louvet C, Mineur L, Bouché O, Chibaudel B, Artru P, Desseigne F, Bachet JB, Mathiot C, Pierga JY, Hammel P ( 2013 ) Circulating tumor cells in locally advanced pancreatic adenocarcinoma: the ancillary CirCe 07 study to the LAP 07 trial . Ann Oncol 24 : 2057 – 2061 .
Brychta N, Krahn T, von Ahsen O ( 2016 ) Detection of KRAS mutations in circulating tumor DNA by Digital PCR in early stages of pancreatic cancer . Clin Chem 62 : 1482 – 1491 .
Chan A, Prassas I, Dimitromanolakis A, Brand RE, Serra S, Diamandis EP, Blasutig IM ( 2014 ) Validation of biomarkers that complement CA19.9 in detecting early pancreatic cancer . Clin Cancer Res 20 : 5787 – 5795 .
DeWitt J, Jowell P, Leblanc J, McHenry L, McGreevy K, Cramer H, Volmar K, Sherman S, Gress F ( 2005 ) EUS-guided FNA of pancreatic metastases: a multicenter experience . Gastrointest Endosc 61 : 689 – 696 .
Diaz LA Jr, Bardelli A ( 2014 ) Liquid biopsies: genotyping circulating tumor DNA . J Clin Oncol 32 : 579 – 586 .
Earl J, Garcia-Nieto S, Martinez-Avila JC, Montans J, Sanjuanbenito A, Rodríguez-Garrote M, Lisa E, Mendía E, Lobo E, Malats N, Carrato A, Guillen-Ponce C ( 2015 ) Circulating tumor cells (Ctc) and kras mutant circulating free Dna (cfdna) detection in peripheral blood as biomarkers in patients diagnosed with exocrine pancreatic cancer . BMC Cancer 15 : 797 .
Eloubeidi MA, Chen VK, Eltoum IA, Jhala D, Chhieng DC, Jhala N, Vickers SM, Wilcox CM ( 2003 a) Endoscopic ultrasound-guided fine needle aspiration biopsy of patients with suspected pancreatic cancer: diagnostic accuracy and acute and 30-day complications . Am J Gastroenterol 98 : 2663 – 2668 .
Eloubeidi MA, Jhala D, Chhieng DC, Chen VK, Eltoum I, Vickers S, Mel Wilcox C, Jhala N ( 2003 b) Yield of endoscopic ultrasound-guided fine-needle aspiration biopsy in patients with suspected pancreatic carcinoma . Cancer 99 : 285 – 292 .
Forbes SA, Beare D, Boutselakis H, Bamford S, Bindal N, Tate J, Cole CG, Ward S, Dawson E, Ponting L, Stefancsik R, Harsha B, Kok CY, Jia M, Jubb H, Sondka Z, Thompson S, De T, Campbell PJ ( 2017 ) COSMIC: somatic cancer genetics at high-resolution . Nucleic Acids Res 45 : D777 – D783 .
Grover PK, Cummins AG, Price TJ, Roberts-Thomson IC, Hardingham JE ( 2014 ) Circulating tumor cells: the evolving concept and the inadequacy of their enrichment by EpCAM-based methodology for basic and clinical cancer research . Ann Oncol 25 : 1506 – 1516 .
Hadano N, Murakami Y, Uemura K, Hashimoto Y, Kondo N, Nakagawa N, Sueda T, Hiyama E ( 2016 ) Prognostic value of circulating tumor DNA in patients undergoing curative resection for pancreatic cancer . Br J Cancer 115 : 59 – 65 .
Harewood GC, Wiersema MJ ( 2002 ) Endosonography-guided fine needle aspiration biopsy in the evaluation of pancreatic masses . Am J Gastroenterol 97 : 1386 – 1391 .
Hofman V, Long E, Ilie M, Bonnetaud C, Vignaud JM, Fléjou JF, Lantuejoul S, Piaton E, Mourad N, Butori C, Selva E, Marquette CH, Poudenx M, Sibon S, Kelhef S, Vénissac N, Jais JP, Mouroux J, Molina TJ, Vielh P, Hofman P ( 2012 ) Morphological analysis of circulating tumor cells in patients undergoing surgery for non-small cell lung carcinoma using the isolation by size of epithelial tumor cell (ISET) method . Cytopathology 23 : 30 – 38 .
Iwanicki-Caron I, Basile P, Toure E, Antonietti M, Lecleire S, Di Fiore A, Oden-Gangloff A, Blanchard F, Lemoine F, Di Fiore F, Sabourin JC, Michel P ( 2013 ) Usefulness of circulating tumor cell detection in pancreatic adenocarcinoma diagnosis . Am J Gastroenterol 108 : 152 – 155 .
Jackson JB, Choi DS, Luketich JD, Pennathur A, Ståhlberg A, Godfrey TE ( 2016 ) Multiplex preamplification of serum DNA to facilitate reliable detection of extremely rare cancer mutations in circulating DNA by digital PCR . J Mol Diagn 18 : 235 – 243 .
Kamisawa T, Wood LD, Itoi T, Takaori K ( 2016 ) Pancreatic cancer . Lancet 388 : 73 – 85 .
Khoja L, Backen A, Sloane R, Menasce L, Ryder D, Krebs M, Board R, Clack G, Hughes A, Blackhall F, Valle JW, Dive C ( 2012 ) A pilot study to explore circulating tumor cells in pancreatic cancer as a novel biomarker . Br J Cancer 106 : 508 – 516 .
Kinugasa H, Nouso K, Miyahara K, Morimoto Y, Dohi C, Tsutsumi K, Kato H, Matsubara T, Okada H, Yamamoto K ( 2015 ) Detection of K-ras gene mutation by liquid biopsy in patients with pancreatic cancer . Cancer 121 : 2271 – 2280 .
Kulemann B, Pitman MB, Liss AS, Valsangkar N, Fernández-Del Castillo C, Lillemoe KD, Hoeppner J, Mino-Kenudson M, Warshaw AL, Thayer SP ( 2015 ) Circulating tumor cells found in patients with localized and advanced pancreatic cancer . Pancreas 44 : 547 – 550 .
Kurihara T, Itoi T, Sofuni A, Itokawa F, Tsuchiya T, Tsuji S, Ishii K, Ikeuchi N, Tsuchida A, Kasuya K, Kawai T, Sakai Y, Moriyasu F ( 2008 ) Detection of circulating tumor cells in patients with pancreatic cancer: a preliminary result . J Hepatobiliary Pancreat Surg 15 : 189 – 195 .
Lamy A, Blanchard F, Le Pessot F, Sesboüé R, Di Fiore F, Bossut J, Fiant E, Frébourg T, Sabourin J-C ( 2011 ) Metastatic colorectal cancer KRAS genotyping in routine practice: results and pitfalls . Mod Pathol 24 : 1090 – 1100 .
LeBlanc JK, Ciaccia D, Al-Assi MT, McGrath K, Imperiale T, Tao L-C, Vallery S, DeWitt J, Sherman S, Collins E ( 2004 ) Optimal number of EUS-guided fine needle passes needed to obtain a correct diagnosis . Gastrointest Endosc 59 : 475 – 481 .
Nouts A, Lévy P, Voitot H, Bernades P ( 1998 ) Diagnostic value of serum Ca 19-9 antigen in chronic pancreatitis and pancreatic adenocarcinoma . Gastroenterol Clin Biol 22 : 152 – 159 .
Poruk KE, Valero V, Saunders T, Blackford AL, Griffin JF, Poling J, Hruban RH, Anders RA, Herman J, Zheng L, Rasheed ZA, Laheru DA, Ahuja N, Weiss MJ, Cameron JL, Goggins M, Iacobuzio-Donahue CA, Wood LD, Wolfgang CL ( 2016 ) Circulating tumor cell phenotype predicts recurrence and survival in pancreatic adenocarcinoma . Ann Surg 264 : 1073 – 1081 .
Sausen M, Phallen J, Adleff V, Jones S, Leary RJ, Barrett MT, Anagnostou V, Parpart-Li S, Murphy D, Kay LiQ, Hruban CA, Scharpf R, White JR, O’Dwyer PJ, Allen PJ, Eshleman JR, Thompson CB, Klimstra DS, Linehan DC, Maitra A, Hruban RH, Diaz LA, Von Hoff DD, Johansen JS, Drebin JA, Velculescu VE ( 2015 ) Clinical implications of genomic alterations in the tumour and circulation of pancreatic cancer patients . Nat Commun 6 : 7686 .
Sefrioui D, Perdrix A, Sarafan-Vasseur N, Dolfus C, Dujon A, Picquenot J-M, Delacour J, Cornic M, Bohers E, Leheurteur M, Rigal O, Tennevet I, Thery J-C, Alexandru C, Guillemet C, Moldovan C, Veyret C, Frebourg T, Di Fiore F, Clatot F ( 2015 a) Short report: Monitoring ESR1 mutations by circulating tumor DNA in aromatase inhibitor resistant metastatic breast cancer . Int J Cancer 137 : 2513 – 2519 .
Sefrioui D, Sarafan-Vasseur N, Beaussire L, Baretti M, Gangloff A, Blanchard F, Clatot F, Sabourin J-C, Sesboüé R, Frebourg T, Michel P, Di Fiore F ( 2015 b) Clinical value of chip-based digital-PCR platform for the detection of circulating DNA in metastatic colorectal cancer . Dig Liver Dis 47 : 884 – 890 .
Singh N, Gupta S, Pandey RM, Chauhan SS, Saraya A ( 2015 ) High levels of cell-free circulating nucleic acids in pancreatic cancer are associated with vascular encasement, metastasis and poor survival . Cancer Invest 33 : 78 – 85 .
Slesak B, Harlozinska-Szmyrka A, Knast W, Sedlaczek P, van Dalen A, Einarsson R ( 2000 ) Tissue polypeptide specific antigen (TPS), a marker for differentiation between pancreatic carcinoma and chronic pancreatitis. A comparative study with CA 19-9 . Cancer 89 : 83 – 88 .
Steinberg W ( 1990 ) The clinical utility of the CA 19-9 tumor-associated antigen . Am J Gastroenterol 85 : 350 – 355 .
Su S-B, Qin S-Y, Chen W, Luo W, Jiang H-X ( 2015 ) Carbohydrate antigen 19-9 for differential diagnosis of pancreatic carcinoma and chronic pancreatitis . World J Gastroenterol 21 : 4323 – 4333 .
Sweeney AD, Wu M-F, Hilsenbeck SG, Brunicardi FC, Fisher WE ( 2009 ) Value of pancreatic resection for cancer metastatic to the pancreas . J Surg Res 156 : 189 – 198 .
Tadic M, Kujundzic M, Stoos-Veic T, Kaic G, Vukelic-Markovic M ( 2008 ) Role of repeated endoscopic ultrasound-guided fine needle aspiration in small solid pancreatic masses with previous indeterminate and negative cytological findings . Dig Dis 26 : 377 – 382 .
Takai E, Totoki Y, Nakamura H, Morizane C, Nara S, Hama N, Suzuki M, Furukawa E, Kato M, Hayashi H, Kohno T, Ueno H, Shimada K, Okusaka T, Nakagama H, Shibata T, Yachida S ( 2015 ) Clinical utility of circulating tumor DNA for molecular assessment in pancreatic cancer . Sci Rep 5 : 18425 .
Uehara H, Ikezawa K, Kawada N, Fukutake N, Katayama K, Takakura R, Takano Y, Ishikawa O, Takenaka A ( 2011 ) Diagnostic accuracy of endoscopic ultrasound-guided fine needle aspiration for suspected pancreatic malignancy in relation to the size of lesions . J Gastroenterol Hepatol 26 : 1256 – 1261 .
Varadarajulu S, Tamhane A, Eloubeidi MA ( 2005 ) Yield of EUS-guided FNA of pancreatic masses in the presence or the absence of chronic pancreatitis . Gastrointest Endosc 62 : 728 – 736 .
Visani M, de Biase D, Baccarini P, Fabbri C, Polifemo AM, Zanini N, Pession A, Tallini G ( 2013 ) Multiple KRAS mutations in pancreatic adenocarcinoma: molecular features of neoplastic clones indicate the selection of divergent populations of tumor cells . Int J Surg Pathol 21 : 546 – 552 .
Wang P, Bahreini A, Gyanchandani R, Lucas P, Hartmaier RJ, Watters RJ, Jonnalagadda AR, Trejo Bittar HE, Berg A, Hamilton RL, Kurland BF, Weiss K, Mathew A, Leone JP, Davidson NE, Nikiforova MN, Brufsky AM, Ambros TF, Stern AM, Puhalla S, Lee AW, Oesterreich S ( 2015 ) Sensitive detection of mono- and polyclonal ESR1 mutations in primary tumors, metastatic lesions and cell free DNA of breast cancer patients . Clin Cancer Res 22 : 1130 – 1137 .
Winter JM, Yeo CJ, Brody JR ( 2013 ) Diagnostic, prognostic, and predictive biomarkers in pancreatic cancer . J Surg Oncol 107 : 15 – 22 .
Yoshinaga S, Suzuki H, Oda I, Saito Y ( 2011 ) Role of endoscopic ultrasound-guided fine needle aspiration (EUS-FNA) for diagnosis of solid pancreatic masses . Dig Endosc 23 (Suppl 1) : S29 – S33 .
Zill OA, Greene C, Sebisanovic D, Siew LM, Leng J, Vu M, Hendifar AE, Wang Z, Atreya CE, Kelley RK, Van Loon K, Ko AH, Tempero MA, Bivona TG, Munster PN, Talasaz A, Collisson EA ( 2015 ) Cell-Free DNA next-generation sequencing in pancreatobiliary carcinomas . Cancer Discov 5 : 1040 – 1048 .
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Sefrioui, D., Blanchard, F., Toure, E. et al. Diagnostic value of CA19.9, circulating tumour DNA and circulating tumour cells in patients with solid pancreatic tumours. Br J Cancer 117, 1017–1025 (2017). https://doi.org/10.1038/bjc.2017.250
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2017.250
Keywords
This article is cited by
-
Liquid biopsy techniques and pancreatic cancer: diagnosis, monitoring, and evaluation
Molecular Cancer (2023)
-
Efficient and accurate KRAS genotyping using digital PCR combined with melting curve analysis for ctDNA from pancreatic cancer patients
Scientific Reports (2023)
-
Liquid biopsy: a step closer to transform diagnosis, prognosis and future of cancer treatments
Molecular Cancer (2022)
-
Prognostic Value of KRAS Mutations in the Peripheral Blood of Patients with Pancreatic Cancer: a Systematic Review and Meta-analysis
Indian Journal of Surgery (2022)
-
Coupled liquid biopsy and bioinformatics for pancreatic cancer early detection and precision prognostication
Molecular Cancer (2021)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}