
Abstract
Background:
Cadmium, an established carcinogen, is a risk factor for prostate cancer. Induction of autophagy is a prerequisite for cadmium-induced transformation and metastasis. The ability of Psoralidin (Pso), a non-toxic, orally bioavailable compound to inhibit cadmium-induced autophagy to prevent prostate cancer was investigated.
Methods:
Psoralidin was studied using cadmium-transformed prostate epithelial cells (CTPE), which exhibit high proliferative, invasive and colony forming abilities. Gene and protein expression were evaluated by qPCR, western blot, immunohistochemistry and immunofluorescence. Xenograft models were used to study the chemopreventive effects in vivo.
Results:
Cadmium-transformed prostate epithelial cells were treated with Pso resulting in growth inhibition, without causing toxicity to normal prostate epithelial cells (RWPE-1). Psoralidin-treatment of CTPE cells inhibited the expression of Placenta Specific 8, a lysosomal protein essential for autophagosome and autolysosome fusion, which resulted in growth inhibition. Additionally, Pso treatment caused decreased expression of pro-survival signalling proteins, NFκB and Bcl2, and increased expression of apoptotic genes. In vivo, Pso effectively suppressed CTPE xenografts growth, without any observable toxicity. Tumours from Pso-treated animals showed decreased autophagic morphology, mesenchymal markers expression and increased epithelial protein expression.
Conclusions:
These results confirm that inhibition of autophagy by Pso plays an important role in the chemoprevention of cadmium-induced prostate carcinogenesis.
Similar content being viewed by others

Main
Cadmium is a non-essential metal, a stable and persistent toxicant that is continuously introduced into the environment through the processing of metal ores and the burning of fossil fuels (Aylett, 1979; Chmielnicka and Cherian, 1986; Friberg et al, 1986; Lewis et al, 1972; Agency for Toxic Substances and Disease Registry, 2012). It is well established that cadmium causes both acute and chronic cytotoxicity. Significant correlations between chronic cadmium exposure and the risk of human colon, breast, lung and prostate cancer have been reported (Kjellstrom et al, 1979; Satarug, 2012; Jones et al, 2016). The precise molecular mechanisms by which this metal causes healthy cells to transform to malignant states however, have yet to be fully defined. Current models propose the induction of reactive oxygen species, impairment of DNA repair, inhibition of p53 signalling, alterations in the Bcl2/Bax ratio and silencing the pro-apoptotic function of caspase cascades may contribute to cadmium-induced prostate carcinogenesis (Chao and Korsmeyer, 1998; Nunez et al, 1998; Achanzar et al, 2002; Aimola et al, 2012).
In vitro studies demonstrated that normal human prostate epithelial cells (RWPE-1) when chronically exposed to cadmium, transformed to malignant cells (cadmium-transformed prostate epithelial; CTPE). The transformation of RWPE-1 cells was confirmed using cellular and molecular markers of cancer phenotypes including adenocarcinoma formation in nude mice (Achanzar et al, 2001; Waalkes, 2003; Benbrahim-Tallaa et al, 2007). Several potential processes that may contribute to cadmium-induced prostate carcinogenesis; including cell proliferation and differentiation, cell cycle progression, DNA synthesis and repair, apoptosis and other cellular activities; have been identified using RWPE-1 and CTPE cells (Beyersmann and Hechtenberg, 1997; Achanzar et al, 2000; Achanzar et al, 2002; Bertin and Averbeck, 2006; Giaginis et al, 2006; Aimola et al, 2012). Additionally, intracellular signalling pathways including mitogen-activated protein kinase, phosphatidylinositol-3-kinase (PI3K), AKT, Nuclear Factor-κB (NFκB), p53 inactivation and induction of epithelial–mesenchymal transition (EMT) may contribute to cadmium-induced prostate carcinogenesis have been characterised using these cell lines (Achanzar et al, 2000, 2001, 2002; Misra et al, 2003; Urani et al, 2014).
The involvement of autophagy, the lysosomal-mediated degradation of damaged organelles, in cadmium-induced carcinogenesis is currently being explored (Luevano and Damodaran, 2014; Son et al, 2014; Cartularo et al, 2016). The induction of autophagy involves many steps to initiate autophagosome formation, which eventually promotes autolysosomal degradation through the activation of the cytosolic microtubule-associated protein 1A/1B-light chain 3A (LC3A) (Hara et al, 2008; Hosokawa et al, 2009; Jung et al, 2009). LC3A can then be converted to LC3B through the processing by Atg7, an E1-like activating enzyme (Wu et al, 2006). Cytosolic LC3B is recruited to the membrane of the phagophore to assist in the formation of the autophagosomes, which are required for induction of cell death. The lysosomal protein Placental Specific 8 (Plac8) is currently being examined as an effector in cellular transformation through autophagy. Plac8 facilitates autophagosome–lysosome fusion by activating pro-survival function of autophagy (Kinsey et al, 2014). Plac8 localises to the lysosomal membrane where it binds to the lysosomal proteins LAMP-1 and -2, which are essential for autolysosomal initiation (Kinsey et al, 2014).
It is well established that cadmium exposure is a risk factor for human prostate carcinogenesis; the second leading cause of cancer death in United States and worldwide (U.S. Cancer Statistics Working Group, 2016). Effectively preventing this disease requires the identification of novel compounds. Natural products have been shown to be chemopreventive candidates for prostate cancer (Wang et al, 2007; Horie, 2012; Aufderklamm et al, 2014; Parnes et al, 2014; Vemana et al, 2014; Kallifatidis et al, 2016). One such product is Psoralidin (Pso), isolated from the seeds of the medicinal pant Psoralea corylifolia (Leguminosae) (Xiao et al, 2010). Psoralidin induces cytotoxicity in various cancers and may have anti-inflammatory, antioxidant and antitumour effects (Mar et al, 2001; Srinivasan et al, 2010; Szliszka et al, 2011; Das et al, 2014; Jin et al, 2016).
In this study, the mechanism by which Pso effectively inhibits oncogenic autophagy and promotes cell death in CTPE cells both in vitro and in vivo was examined. Our results demonstrated that Pso inhibited autophagy flux by Plac8 inhibition and induce apoptosis. In addition, oral administration of Pso inhibited tumour growth of CTPE cells in mice, suggesting that autophagy inhibition may prevent prostate cancer growth.
Materials and methods
Cell lines and reagents
The normal prostate epithelial cell line RWPE-1 was purchased from the American Type Culture Collection (Manassas, VA, USA). cadmium-transformed prostate epithelial cells were generous gift from Dr Mike Waalkes (NIEHS). Both cell lines were maintained in KSFM medium containing 50 μg ml−1 bovine pituitary extract, 5 ng ml−1 epidermal growth factor and 1% antibiotic and antimycotic solution in a humidified atmosphere of 5% CO2 at 37 °C. 10 μM cadmium (final concentration) was used for in vitro studies. The following antibodies were obtained from Cell Signaling Technology (Danvers, MA, USA): cell survival (PI3K p85, p110, p65, Bcl-2), autophagy markers (Plac8, Lamp-1, LC3B, Atg-3, -5, -7, 12, 16L, Beclin) and apoptosis markers (BAX, cleaved caspase-9, -3 and PARP). Beta-actin, Lamin A/C, anti-mouse, anti-goat and anti-rabbit secondary antibodies conjugated with HRP were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). TUNEL kits were purchased from BD Biosciences (San Diego, CA, USA). Alexa Fluor 488 and prolong gold anti-fade with DAPI mount were purchased from Invitrogen (Grand Island, NY, USA).
Cell proliferation and soft agar colony formation assays
Cells were treated with various concentrations of Pso in DMSO for 24 h. Cell viability was then measured using MTT assays (Cell Signaling, Danvers, MA, USA), according to the manufacturer's protocol and as previously described (Srinivasan et al, 2010; Roy et al, 2013; Suman et al, 2013, 2014a, 2014b). Anchorage-independent growth was monitored by colony formation assay using a CytoSelectTM 96-well in vitro tumour sensitivity assay kit (Cell Biolabs, Inc., San Diego, CA, USA), per the manufacturer’s instructions (Suman et al, 2014a).
Apoptosis assay
Apoptosis assays were performed using terminal deoxynucleotidyl transferase dUTP nick end labelling TUNEL Apoptosis Detection Kits (EMD Millipore, Billerica, MA, USA) as per the manufacture protocol. Briefly, cells were grown on coverslips and then treated with Pso (4 μM) for 24 h. Following this incubation, cells were fixed with paraformaldehyde for 1 h, then permeabilised with 0.1% Triton X-100 and incubated with 50 μl TUNEL reaction mixture. The presence and number of apoptotic cells were determined by confocal microscopy.
Protein extraction and western blotting
The CTPE cells were seeded in 6-well plates. After 24 h, cells were incubated with Pso (4 μM) or DMSO (vehicle control) for 12 and 24 h. Whole protein lysates were then prepared using Mammalian Protein Extraction Reagent (Thermo Scientific) according to the manufacturer’s protocol. Following gel electrophoresis, western blotting was performed using antibodies against P13K, p65, Plac8, Lamp-1, LC3B, Atg-3, -5, -7, 12, 16L, Beclin, Lamin A cleaved caspase-9, -3 and PARP. Levels of protein expression were measured by chemiluminescence (Roy et al, 2013; Suman et al, 2013, 2014a, 2014b). The NIH image J software was used for densitometry analysis.
Real time quantitative PCR
Total RNA was isolated from vehicle and Pso-treated CTPE cells by Qiagen, RNeasy Kit and 1 μg RNA was used for cDNA synthesis using the Applied Biosystems cDNA synthesis kit by using SYBR Green super mix (QIAGEN Inc., Valencia, CA, USA), Quantitative RT-PCR was performed as previously published.
NFκB activation assay
To measure NFκB activation in control and Pso-treated CTPE cells, nuclear extracts were prepared using N-PER-Nuclear and Cytoplasmic Extraction Reagents (Thermo Scientific) according to the manufacturer’s protocol. The NFκB p65 Transcription Factor Assay Kit (Abcam, Cambridge, MA, USA) was used to detect NFκB activation according to the manufacturer’s protocol.
Invasion assays
The effects of Pso on the invasive properties were determined using Boyden chambers equipped with 8-μm pore size polyethylene terephthalate membranes coated with Matrigel (BD Biosciences, San Jose, CA, USA). Approximately 2 × 104 cells in media with or without Pso were layered over the gel and allowed to migrate for 24. Cells were then stained, visualised and counted.
Wound healing assay
Pso-containing medium was added to cells plated in 6-well plates and after reaching confluency. Wounds were generated by scratching with a pipette tip at the base of each well plate. Images were acquired at 0 h and 24 h after wound creation. The distance that cells migrated through the area created by scratching was determined by using NIS-Element AR software (Nikon Instruments Inc., Melville, NY, USA)
Immunofluorescence staining
Cadmium-transformed prostate epithelial cells were seeded onto coverslips and allowed to attach for 24 h. The cells were treated for 24 h with Pso and then fixed and permeabilised. Cells were incubated with primary antibodies to Plac8, LAMP1 or LC3B overnight. After washing, cells were incubated with Alexa Fluor 488- or 594-labelled secondary antibodies for 45 min. and counterstained with DAPI to label nuclei. The presence and locations of Plac8, LAMP1 and LC3B were visualised using immunofluorescence microscopy, as previously described (Roy et al, 2013; Suman et al, 2013, 2014a, 2014b).
Xenograft studies
All animals were housed under pathogen-free conditions, and experiments were performed in accordance with Institutional Animal Care & Use Committee (IACUC) and approved by University of Louisville. Balb/c athymic nude mice (nu/nu) were purchased from The Jackson Laboratory and used at 6 weeks of age. For subcutaneous tumour xenograft studies, CTPE cells approximately (1.5 × 105) in 50-μl (final volume) of phosphate-buffered saline and Matrigel (1 : 1) were injected into separate flanks of mice (n=10). Mice were monitored twice weekly and tumour volumes were measured once a week. To measure the ability of Pso to inhibit tumour cell growth, Pso (20 mg kg−1) was given orally for 5 days for 4–5 weeks. Control mice were received sesame oil.
For histological examination, tumour samples derived from CTPE and Pso-treated tumour xenografts were examined using light microscopy following fixation staining with hematoxylin and eosin (H&E). Similarly, tumour sections were stained with primary antibodies for Plac8, β-catenin and Ki-67 followed by incubation with secondary antibody.
Statistical analysis
Statistical analyses were performed using GraphPad Prism 5.0 (GraphPad Software, Inc., La Jolla, CA, USA) and SPSS 15.0 software (IBM SPSS Statistics, IBM, Ehningen, Germany). Differences between treatments groups were analysed either using a two-sample Student’s t-test for normally distributed variables or the Wilcoxon rank sum test for non-normally distributed variables or one way ANOVA. P values below 0.05 were considered to be significant. Data are presented as the mean±s.d. of a minimum of three independent experiments unless stated otherwise. Significant difference from control values was indicated at *P=0.05, **P=0.01 and ***P=0.001
Results
Psoralidin inhibits CTPE cell growth and induces apoptosis
We explored the anticancer effects of Pso in CTPE cells. To measure levels of cytotoxicity, cells were treated with different concentration of Pso (0–10 μM) for 24 h Psoralidin inhibited CTPE cell growth and proliferation with an IC50 value of ∼4 μM. Conversely, Pso did not significantly inhibit normal prostate epithelial cell growth (Figure 1). These results indicate that Pso specifically targets transformed prostate epithelial cells. The ability of Pso to inhibit CTPE cell growth in soft agar, which is a stringent test for malignant cell transformation, was also determined. Exposure to Pso at concentrations >4 μM significantly (P<0.01) inhibited the colony forming ability of CTPE cells by almost 2-fold, as compared with vehicle (Figure 1).

Psoralidin cytotoxicity in RWPE-1 and CTPE cells. (upper panel) RWPE-1 and CTPE cells were treated with vehicle (DMSO; square) or the indicated concentrations of Pso (circle) for 24 h and cell viability determined. (middle panel) Cell proliferation was assessed by BrdU incorporation in CTPE cells treated with indicated concentrations of Pso. Data are expressed as mean±s.d. of two independent experiments done in triplicate. (lower panels) To assess anchorage-independent growth, CTPE (1 × 103) cells were grown in soft agar with or without Pso for 10–15 days. Colonies were stained with crystal violet and counted manually. Student’s t-test was used to calculate statistical significance between vehicle control and treatment at each concentration. **P<0.01, ***P<0.001, ****P<0.0001.
The effect of psoralidin on autophagy in CTPE cells
Low concentrations of cadmium induce autophagy-promoted cell proliferation in non-transformed cells (Dong et al, 2009). To determine if the growth inhibitory effects of Pso in CTPE cells is mediated by autophagy, changes in protein levels of autophagy markers were measured. In Pso-treated CTPE cells, the steady-state levels of Beclin, Atg-3, -5, 7–12 and -16L increased. The levels of LC3B increased with concomitant time dependent decreases in the expression of Plac8 and LAMP1 (Figure 2). Psoralidin treatment of CTPE cells lead to significant changes in LC3B and Plac8 mRNA levels, similar to those observed for protein (Figure 2). A significant difference in LAMP1 mRNA levels was not observed.

Psoralidin-induced autophagy in CTPE cells. (upper left panel) Whole-cell lysates were prepared for western blot analysis to determine the expression levels of LC3IB and IIB, Plac8, LAMP1, Beclin, Atg-3, Atg-5, Atg-7, Atg-12 and Atg16L. β-actin was used as a loading control. (lower left panel) Total RNA was extracted and qRT-PCR performed to measure differences between the steady-state levels of expression of LC3B, Lamp1 and Plac8 from Pso-treated and -untreated CTPE cells. (upper right panel) Confocal microscopy was used with CTPE cells immunostained with Plac8, LAMP1 and LC3B antibodies. Autophagosome formation is marked by the presence of cytoplasmic puncta. (lower right panel) Localisation of Plac8 and LC3B; Cells treated in presence or absence of Pso and immunostained for Plac8 detection (green- Alexfluor 488) to visualise the overlap with LC3B (Red-alexafluor 594) which is shown as yellow. Student’s t-test was used to calculate statistical significance between vehicle control and treatment at each concentration. *P<0.05 and ***P<0.001. Densitometric analysis for western blots from two or three experiments are given in Supplementary Information.
Immunofluorescence analysis was used to define changes in the sub-cellular localisation of Plac8, LAMP1 and LC3B associated with Pso treatment in CTPE cells. In untreated cells, low levels of LC3B was observed in cytosol (Figure 2). Following Pso treatment, the level of Plac8 mRNA decreased similar to the cognate protein level. In contrast, the levels of LC3B appeared as perinuclear puncta, likely associated with autolysosomes and autophagosomes, respectively (Figure 2). To confirm inhibition of Plac8 blocks the fusion of autophagosome and autolysosome in CTPE cells, we analysed the co-localisation of Plac8 and LC3B in controls, as wells CTPE cells treated with Pso. Control cells exhibited a co-localisation of Plac8 and LC3B, however inhibition of Plac8 prevented autophagosome (Plac8) and autolysosome (LC3B) fusion in Pso-treated CTPE cells which was similar to chloroquine treatment (Figure 2).
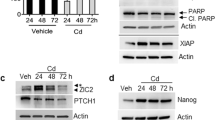
TUNEL assays were performed to determine if the inhibition of cell growth by Pso was due to an induction of apoptosis. Treatment of CTPE cells with 4 μM Pso for 24 h resulted in a significant (P<0.001) increase in the number of TUNEL positive cells, compared to vehicle-treated cells. In addition, time-dependent increases in the levels of the pro-apoptotic BAX protein and caspase cascade proteins; cleaved caspase-9, caspase-3, and poly-ADP ribose polymerase (PARP) were observed in Pso-treated CTPE cells (Figure 3).

Psoralidin-induced apoptosis in CTPE cells. CTPE cells were treated with 4 μM Pso for 12 and 24 h and the levels of Bax, cleaved caspase-9, caspase-3 and PARP were measured by western blotting. Densitometric analysis of western blots from one, two or three experiments are given in Supplementary Information. β-actin was used as a loading control. CTPE cells were treated either with vehicle or Pso for 24 h and fixed with paraformaldehyde and permeabilised with sodium citrate and Triton X. DNA fragmentation was determined by TdT-mediated dUTP nick end labelling (TUNEL) (red channel). DAPI (blue channel) was used to locate the nuclei of the cell and numbers of cells were counted manually. Student’s t-test was used to calculate statistical significance between vehicle control and Pso treatment. **P<0.01
Psoralidin inhibits NFκB signalling in CTPE cells
Cadmium induces the activity of pro-survival factors, such as NFκB and AP-1 in both cultured cells and animal models (Yang et al, 2007; Freitas and Fernandes, 2011). The effects of Pso on NFκB and its downstream targets in CTPE cells were determined. Pso significantly inhibited (P<0.05) NFκB binding to its cognate promoter element. There were also significant, time-dependent decreases in the expression of the downstream effector, the anti-apoptotic protein Bcl-2 (Figure 4A). These results were further confirmed by analysing cytosolic and nuclear fraction of CTPE cells after Pso treatment: downregulation of p65 expression and NFκB activation were seen in the nuclear fraction of CTPE cells (Figure 4B and C). While analysing upstream of NFκB regulators, Pso treatment inhibited the expression of p85, a catalytic unit of PI3K complex without affecting the total levels of p110 (Figure 4A).

Effects of Psoralidin on NF κ B signalling in CTPE cells. (A) Western blot analyses of PI3K/NFκB signalling in untreated and Pso-treated CTPE cells. Densitometric analysis of western blots from two or three experiments are given in Supplementary Information. (B) Nuclear extract was prepared from cells exposed to 4 μM Pso for 24 h and untreated control cells. The levels of p65 in nuclear and cytoplasmic fractions were determined by western blot using Lamin A and β-actin as loading control, respectively. (C) Nuclear factions were subjected NFκB p65 Transcription Factor Assay by EMSA analysis. (D) CTPE cells were untreated or exposed to 4 μM Pso for 12 and 24 h. The levels of E-cadherin, β-catenin, Vimentin and MMP9 measured by western blot analysis with β-actin used as a loading control. (E) qRT-PCR was used to measure the steady-state mRNA levels of E-cadherin, β-catenin and MMP9 in untreated CTPE cells and those treated with Pso for 12 and 24 h. Densitometric analysis of western blots from two or three experiments are given in Supplementary Information. Student’s t-test was used to calculate statistical significance between vehicle control (UT) and Pso-treated cells at each time point. *P<0.05, **P<0.01, ***P<0.001.
Inhibition of EMT phenotypes in CTPE cells by Pso
Earlier studies demonstrated that CTPE cells express increased levels of secreted matrix metalloproteinase-2 (MMP2) and MMP9, which correlated with the invasive phenotype in xenograft models (Achanzar et al, 2001; Son et al, 2012). The effect of Pso on EMT phenotypes, invasion and migration, were determined in CTPE cells. Pso significantly (P<0.0001) inhibited invasion of CTPE cells by 2.5-fold, compared to the vehicle-treated control cells using a trans-well invasion assay. In a wound healing migration assay, vehicle-treated CTPE cells exhibited complete wound recovery (∼30%) after 24 h, whereas Pso treatment significantly (P<0.05) inhibited the migration of CTPE cells to 4.6%. The effect of 12 and 24 h Pso treatment on the steady-state levels of EMT proteins E-cadherin, β-catenin, Vimentin and MMP9 in CTPE cells were determined by western blot analysis. A time dependent, significant induction of E-cadherin (four-fold) was observed. In contrast, Pso treatment caused significant decreases in the expression of β-catenin, Vimentin, and MMP9 (Figure 4D). The fold change in the steady-state mRNA levels of EMT genes was determined using qRT-PCR. Similar to the protein results, significant increases in E-cadherin with decreases in MMP9 expression were observed following Pso-treatment (Figure 4E). The levels of β-catenin mRNA did not significantly change. Combined these results suggest that Pso may be a potent inhibitor of metastasis.
Oral administration of Pso inhibited CTPE tumour xenograft formation in nude mice
We examined the anti-cancer ability of Pso in vivo using the xenograft model. Oral administration of Pso significantly inhibited the tumour growth, compared to vehicle control-treated animals (Figure 5). No adverse toxic effects were found in any organs of Pso-treated animals and body weights were similar to vehicle-treated mice (Figure 5).

Psoralidin inhibits CTPE xenografts tumour growth in nude mice. CTPE cells in a 50-μl (final volume) of phosphate-buffered saline and Matrigel (1 : 1) were subcutaneously injected into separate flanks of mice. Mice received vehicle (sesame oil; circles) or Pso (20 mg kg−1 Bw; squares) orally for 5 days per week up to 5 weeks. Tumour volumes (mm3) (upper panel) and mouse body weight (lower panel) were measured twice and once a week, respectively. Student’s t-test was used to calculate statistical significance between Pso-treated and -untreated animals at each time point. *P<0.05, **P<0.01, ***P<0.001.
Histological analysis, H&E staining, of tumours from control and Pso-treated mice showed distinct nuclear and cytosolic components with high intensity in CTPE tumour tissues and induction a high grade mesenchymal component that represented an EMT (Figure 6). Immunohistochemical examination revealed decreased expression of Plac8 in Pso-treated tumour sections. Decreased numbers of cells expressing the EMT marker β-catenin were also seen in Pso-treated animal. A high expression of the cell proliferation marker Ki-67 was observed in sections derived from CTPE tumours from control mice, whereas a decrease in Ki-67 expression was observed Pso-treated animals (Figure 6, Supplementary Table 1). Reticulocyte staining (angiogenesis marker) revealed increased microvessel formation, indicative of the aggressiveness of CTPE tumour growth in vehicle-treated animals, which was not observed in mice receiving Pso.

(upper panel) Vehicle and Pso-treated tumour tissues were processed for H &E and immunohistochemical staining for Plac8, β -catenin, Ki-67 and RETIC. Total of 300–400 cells were counted for positive cells and represented in Supplementary Table 1 as average number of positive cells. (lower panel) qRT-PCR was performed to measure differences between the steady-state levels of expression of EMT markers in CTPE tumours from Pso-treated and -untreated animals. Student’s t-test was used to calculate statistical significance between Pso-treated and -untreated tumours at each time point *P<0.05, **P<0.01, ***P<0.001.
Pso treatment caused decreased steady-state mRNA levels of the EMT makers MMP2, MMP9, Snail and β-catenin with a significant increase (P=0.001) in the epithelial marker E-cadherin (Figure 6). These results support a role for Pso as a potent compound that inhibits tumour growth.
Discussion
Cadmium is a known human carcinogen and long-term exposure is associated with many cancer types (Kjellstrom et al, 1979; Takenaka et al, 1983; Waalkes et al, 1992; Julin et al, 2012; Satarug, 2012; Agency for Toxic Substances and Disease Registry, 2015; International Programme on Chemical Safety, 2015). In the present study, the chemopreventive effect of Pso, as demonstrated by growth inhibition of CTPE cells in vitro and in vivo, was shown. The molecular mechanism by which Pso inhibits cadmium-transform prostate cells is associated with inhibition of Plac8, which is responsible for autophagosome and autolysosome fusion and induction of apoptosis.
Consistent with previous results, CTPE cells grow aggressively, compared to the non-transformed parental RWPE-1 cell line, and exhibit malignant phenotypes such as anchor independent growth and mesenchymal characteristics (Chakraborty et al, 2010). At a non-cytotoxic concentration, Pso significantly inhibited the growth and colony forming ability of CTPE cells (Figure 1). In addition, induction of TUNEL positive cells in Pso-treated CTPE cells suggesting involvement of DNA-damage or fragmentation leading to cell death in CTPE cells.
Published evidence suggests that low concentrations of cadmium induce autophagy-promoted cell proliferation and that higher concentrations induce cell death (Dong et al, 2009). The induction of autophagy-mediated survival mechanism due to lysosome genes (Turk and Turk, 2009). Recently, (Santanam et al, 2016) reported that deletion of Atg7 in Pten-deficient mice ((Nkx3.1CreERT2/+;PtenF/F; AtgΔ/Δ) caused delayed prostate tumour progression, as well as castration resistant prostate cancer, suggesting that autophagy could be a therapeutic target for prostate cancer. Our findings on global gene arrays showed that the lysosome gene, Plac8 is highly expressed in CTPE cells, compared to RWPE-1 cells (data not shown). In addition, the induction of Plac8 in CTPE cells is responsible for autophagosome/autolysosome fusion that results in cell survival and proliferation. Inhibition of Plac8 by Pso blocks autophagosome/autolysosome fusion which leads to cell death in CTPE cells. Plac8 functions differ in each cell type; overexpression of Plac8 induces apoptosis in human lymphocytes, whereas in fibroblasts it protects cells (Rogulski et al, 2005; Mourtada-Maarabouni et al, 2013). Plac8 induces EMT in colon cancer cells and cell cycle regulation in pancreatic cancer cells (Li et al, 2014; Kaistha et al, 2016). Plac8 also facilitates autophagosome–lysosome fusion by activating the pro-survival function of autophagy in pancreatic cancer (Kinsey et al, 2014). Plac8 localises to the lysosomal membrane where it binds to the lysosomal proteins Lamp-1 and -2, which are essential for autosomal initiation (Kinsey et al, 2014). Similarly, in our results downregulation of Plac8 by Pso in cadmium-transformed cells resulted in inhibition of growth in both cell culture and xenograft model of CTPE. In our results the inhibition of Plac8 prevented autophagosome and autolysosome fusion although induction of LC3B activation (RNA, protein and LC3b puncta) were seen in Pso-treated cells. Inhibition of autophagy resulted in induction of cell death in Pso-treated CTPE cells, which is similar to previous findings that accumulation of autophagy vacuoles triggers induction of cell death (apoptosis) without altering mitochondrial membrane (Boya et al, 2005). To support apoptotic induction, we have seen TUNEL positive cells in Pso-treated cells, also confirmed the activation of Caspases and PARP cleavage in CTPE cells suggesting inhibition of autophagy facilitates pro-apoptotic machinery in CTPE cells.
Recently, Son et al demonstrated that induction of reactive oxygen species (ROS)-activated PI3K/AKT signalling in cadmium-transformed normal lung epithelial cells (BEAS-2B) (Son et al, 2012). In our studies, inhibition of NFκB activation and Bcl-2 expression were observed. NFκB activation has been reported in cadmium-transformed cells (Souza et al, 2004; Yang et al, 2007; Freitas and Fernandes, 2011) and silencing NFκB activation triggers induction of apoptosis in cadmium-treated cells (Xie and Shaikh, 2006). Similarly, inhibition of NFκB/Bcl2 and concomitant induction of pro-apoptotic genes BAX and caspase signalling-induced cell death in Pso-treated CTPE cells. The link between PI3K/AKT signalling and oncogenic autophagy is not well defined; however, inhibition of Plac8 and NFκB signalling facilitated cell death in our studies.
We observed that CTPE cells exhibit mesenchymal characteristics and higher constitutive levels of mesenchymal markers, β-catenin, MMP2, MMP9 and Snail both in vivo and in vitro. It’s been reported that a higher levels of mesenchymal markers in different cell types treated with cadmium (Chakraborty et al, 2010; Son et al, 2012, 2014). H&E staining from vehicle-treated CTPE tumours exhibited a higher mesenchymal characteristic, and CTPE cells are highly invasive, based on cell migration and wound healing assays (Supplementary Figure 1). These results correlated with our earlier findings that prostate cancer cells exhibit a higher expression of mesenchymal markers such as β-catenin, snail and Slug; a hallmark for aggressive phenotype of prostate cancer (Das et al, 2014; Suman et al, 2016). Similar molecular kinetics were found in CTPE cells and downregulation of mesenchymal genes by Pso were observed in both western blot and real time PCR analyses. Together, these results demonstrate that Pso effectively prevents CTPE tumour growth, suggesting the feasibility of preventing cadmium-induced prostate cancer by this natural compound. Pso is not only the compound that inhibits CTPE tumour formation, several dietary agents including ECGC (Khan and Mukhtar, 2010), Resveratrol (Palozza et al, 2010) Lycopene (Giovannucci et al, 1995) and diallyl disulphide (Gunadharini et al, 2006) effectively inhibit prostate cancer growth in both pre-clinical and clinical settings.
Confirming in vitro findings in animal studies suggest that inhibition of autophagy could be the mechanism of action for prevention of cadmium-induced prostate cancer. Inhibition of NFκB and Plac8 suggests that these might be key players for the proliferation of CTPE cells. In conclusion, our results revealed an important insight in Plac8-mediated oncogenic autophagy-mediated cellular functions. Inhibition of the Plac8 signalling axis could be a potential biomarker or potential target for cadmium-exposed workers. However, more in depth studies may be required to understand the oncogenic function of Plac8 in prostate cancer.
Change history
27 June 2017
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Achanzar WE, Achanzar KB, Lewis JG, Webber MM, Waalkes MP (2000) Cadmium induces c-myc, p53, and c-jun expression in normal human prostate epithelial cells as a prelude to apoptosis. Toxicol Appl Pharmacol 164: 291–300.
Achanzar WE, Diwan BA, Liu J, Quader ST, Webber MM, Waalkes MP (2001) Cadmium-induced malignant transformation of human prostate epithelial cells. Cancer Res 61: 455–458.
Achanzar WE, Webber MM, Waalkes MP (2002) Altered apoptotic gene expression and acquired apoptotic resistance in cadmium-transformed human prostate epithelial cells. Prostate 52: 236–244.
Agency for Toxic Substances and Disease Registry (2012) Toxicological Profile for Cadmium (update). Agency for Toxic Substances and Disease Registry: Atlanta, GA, USA.
Agency for Toxic Substances and Disease Registry (2015) CERCLA Priority List of Hazardous Substances. Agency for Toxic Substances and Disease Registry: Atlanta, GA, USA.
Aimola P, Carmignani M, Volpe AR, Di Benedetto A, Claudio L, Waalkes MP, van Bokhoven A, Tokar EJ, Claudio PP (2012) Cadmium induces p53-dependent apoptosis in human prostate epithelial cells. PLoS One 7: e33647.
Aufderklamm S, Miller F, Galasso A, Stenzl A, Gakis G (2014) Chemoprevention of prostate cancer by isoflavonoids. Recent Results Cancer Res 202: 101–108.
Aylett BJ (1979) The Chemistry, Biochemistry and Biology of Cadmium. In: Webb M (ed). Elsevier/North-Holland: New York, NY, USA, pp 1–62.
Benbrahim-Tallaa L, Waterland RA, Dill AL, Webber MM, Waalkes MP (2007) Tumor suppressor gene inactivation during cadmium-induced malignant transformation of human prostate cells correlates with overexpression of de novo DNA methyltransferase. Environ Health Perspect 115: 1454–1459.
Bertin G, Averbeck D (2006) Cadmium: cellular effects, modifications of biomolecules, modulation of DNA repair and genotoxic consequences (a review). Biochimie 88: 1549–1559.
Beyersmann D, Hechtenberg S (1997) Cadmium, gene regulation, and cellular signalling in mammalian cells. Toxicol Appl Pharmacol 144: 247–261.
Boya P, Gonzalez-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, Metivier D, Meley D, Souquere S, Yoshimori T, Pierron G, Codogno P, Kroemer G (2005) Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol 25: 1025–1040.
Cartularo L, Kluz T, Cohen L, Shen SS, Costa M (2016) Molecular mechanisms of malignant transformation by low dose cadmium in normal human bronchial epithelial cells. PLoS One 11: e0155002.
Chakraborty PK, Scharner B, Jurasovic J, Messner B, Bernhard D, Thevenod F (2010) Chronic cadmium exposure induces transcriptional activation of the Wnt pathway and upregulation of epithelial-to-mesenchymal transition markers in mouse kidney. Toxicol Lett 198: 69–76.
Chao DT, Korsmeyer SJ (1998) BCL-2 family: regulators of cell death. Annu Rev Immunol 16: 395–419.
Chmielnicka J, Cherian MG (1986) Environmental exposure to cadmium and factors affecting trance-metal metabolism and metal toxicity. Biol Trace Elem Res 10: 243–256.
Das TP, Suman S, Damodaran C (2014) Induction of reactive oxygen species generation inhibits epithelial-mesenchymal transition and promotes growth arrest in prostate cancer cells. Mol Carcinog 53: 537–547.
Dong Z, Wang L, Xu J, Li Y, Zhang Y, Zhang S, Miao J (2009) Promotion of autophagy and inhibition of apoptosis by low concentrations of cadmium in vascular endothelial cells. Toxicol In Vitro 23: 105–110.
Freitas M, Fernandes E (2011) Zinc, cadmium and nickel increase the activation of NF-kappaB and the release of cytokines from THP-1 monocytic cells. Metallomics 3: 1238–1243.
Friberg L, Kjellstrom T, Nordberg GF (1986) In: Friberg L, Nordberg GF, Vouk V (eds). Handbook of the Toxicology of Metals. Elsevier/North-Holland: Amsterdam, The Netherlands, pp 130–237.
Giaginis C, Gatzidou E, Theocharis S (2006) DNA repair systems as targets of cadmium toxicity. Toxicol Appl Pharmacol 213: 282–290.
Giovannucci E, Rimm EB, Ascherio A, Stampfer MJ, Colditz GA, Willett WC (1995) Alcohol, low-methionine—low-folate diets, and risk of colon cancer in men. J Natl Cancer Inst 87: 265–273.
Gunadharini DN, Arunkumar A, Krishnamoorthy G, Muthuvel R, Vijayababu MR, Kanagaraj P, Srinivasan N, Aruldhas MM, Arunakaran J (2006) Antiproliferative effect of diallyl disulfide (DADS) on prostate cancer cell line LNCaP. Cell Biochem Funct 24: 407–412.
Hara T, Takamura A, Kishi C, Iemura S, Natsume T, Guan JL, Mizushima N (2008) FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol 181: 497–510.
Horie S (2012) Chemoprevention of prostate cancer: soy isoflavones and curcumin. Korean J Urol 53: 665–672.
Hosokawa N, Sasaki T, Iemura S, Natsume T, Hara T, Mizushima N (2009) Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy 5: 973–979.
International Programme on Chemical Safety (2015) Cadmium. World Health Organization: Geneva, Switzerland.
Jin Z, Yan W, Jin H, Ge C, Xu Y (2016) Psoralidin inhibits proliferation and enhances apoptosis of human esophageal carcinoma cells via NF-kappaB and PI3K/Akt signaling pathways. Oncol Lett 12: 971–976.
Jones MR, Joshu CE, Kanarek N, Navas-Acien A, Richardson KA, Platz EA (2016) Cigarette smoking and prostate cancer mortality in four US states, 1999-2010. Prev Chronic Dis 13: E51.
Julin B, Wolk A, Bergkvist L, Bottai M, Akesson A (2012) Dietary cadmium exposure and risk of postmenopausal breast cancer: a population-based prospective cohort study. Cancer Res 72: 1459–1466.
Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, Kundu M, Kim DH (2009) ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell 20: 1992–2003.
Kaistha BP, Lorenz H, Schmidt H, Sipos B, Pawlak M, Gierke B, Kreider R, Lankat-Buttgereit B, Sauer M, Fiedler L, Krattenmacher A, Geisel B, Kraus JM, Frese KK, Kelkenberg S, Giese NA, Kestler HA, Gress TM, Buchholz M (2016) PLAC8 localizes to the inner plasma membrane of pancreatic cancer cells and regulates cell growth and disease progression through critical cell-cycle regulatory pathways. Cancer Res 76: 96–107.
Kallifatidis G, Hoy JJ, Lokeshwar BL (2016) Bioactive natural products for chemoprevention and treatment of castration-resistant prostate cancer. Semin Cancer Biol 40-41: 160–169.
Khan N, Mukhtar H (2010) Cancer and metastasis: prevention and treatment by green tea. Cancer Metastasis Rev 29: 435–445.
Kinsey C, Balakrishnan V, O'Dell MR, Huang JL, Newman L, Whitney-Miller CL, Hezel AF, Land H (2014) Plac8 links oncogenic mutations to regulation of autophagy and is critical to pancreatic cancer progression. Cell Rep 7: 1143–1155.
Kjellstrom T, Friberg L, Rahnster B (1979) Mortality and cancer morbidity among cadmium-exposed workers. Environ Health Perspect 28: 199–204.
Lewis GP, Coughlin LL, Jusko WJ, Hartz S (1972) Contribution of cigarette smoking to cadmium accumulation in man. Lancet 1: 291–292.
Li C, Ma H, Wang Y, Cao Z, Graves-Deal R, Powell AE, Starchenko A, Ayers GD, Washington MK, Kamath V, Desai K, Gerdes MJ, Solnica-Krezel L, Coffey RJ (2014) Excess PLAC8 promotes an unconventional ERK2-dependent EMT in colon cancer. J Clin Invest 124: 2172–2187.
Luevano J, Damodaran C (2014) A review of molecular events of cadmium-induced carcinogenesis. J Environ Pathol Toxicol Oncol 33: 183–194.
Mar W, Je KH, Seo EK (2001) Cytotoxic constituents of Psoralea corylifolia. Arch Pharm Res 24: 211–213.
Misra UK, Gawdi G, Pizzo SV (2003) Induction of mitogenic signalling in the 1LN prostate cell line on exposure to submicromolar concentrations of cadmium. Cell Signal 15: 1059–1070.
Mourtada-Maarabouni M, Watson D, Munir M, Farzaneh F, Williams GT (2013) Apoptosis suppression by candidate oncogene PLAC8 is reversed in other cell types. Curr Cancer Drug Targets 13: 80–91.
Nunez G, Benedict MA, Hu Y, Inohara N (1998) Caspases: the proteases of the apoptotic pathway. Oncogene 17: 3237–3245.
Palozza P, Colangelo M, Simone R, Catalano A, Boninsegna A, Lanza P, Monego G, Ranelletti FO (2010) Lycopene induces cell growth inhibition by altering mevalonate pathway and Ras signaling in cancer cell lines. Carcinogenesis 31: 1813–1821.
Parnes HL, Brawley OW, Minasian LM, Ford LG (2014) Phase III prostate cancer chemoprevention trials. Recent Results Cancer Res 202: 73–77.
Rogulski K, Li Y, Rothermund K, Pu L, Watkins S, Yi F, Prochownik EV (2005) Onzin, a c-Myc-repressed target, promotes survival and transformation by modulating the Akt-Mdm2-p53 pathway. Oncogene 24: 7524–7541.
Roy RV, Suman S, Das TP, Luevano JE, Damodaran C (2013) Withaferin A, a steroidal lactone from Withania somnifera, induces mitotic catastrophe and growth arrest in prostate cancer cells. J Nat Prod 76: 1909–1915.
Santanam U, Banach-Petrosky W, Abate-Shen C, Shen MM, White E, DiPaola RS (2016) Atg7 cooperates with Pten loss to drive prostate cancer tumor growth. Genes Dev 30: 399–407.
Satarug S (2012) Long-term exposure to cadmium in food and cigarette smoke, liver effects and hepatocellular carcinoma. Curr Drug Metab 13: 257–271.
Son YO, Pratheeshkumar P, Roy RV, Hitron JA, Wang L, Zhang Z, Shi X (2014) Nrf2/p62 signaling in apoptosis resistance and its role in cadmium-induced carcinogenesis. J Biol Chem 289: 28660–28675.
Son YO, Wang L, Poyil P, Budhraja A, Hitron JA, Zhang Z, Lee JC, Shi X (2012) Cadmium induces carcinogenesis in BEAS-2B cells through ROS-dependent activation of PI3K/AKT/GSK-3beta/beta-catenin signaling. Toxicol Appl Pharmacol 264: 153–160.
Souza V, Escobar Md Mdel C, Gomez-Quiroz L, Bucio L, Hernandez E, Cossio EC, Gutierrez-Ruiz MC (2004) Acute cadmium exposure enhances AP-1 DNA binding and induces cytokines expression and heat shock protein 70 in HepG2 cells. Toxicology 197: 213–228.
Srinivasan S, Kumar R, Koduru S, Chandramouli A, Damodaran C (2010) Inhibiting TNF-mediated signaling: a novel therapeutic paradigm for androgen independent prostate cancer. Apoptosis 15: 153–161.
Suman S, Das TP, Damodaran C (2013) Silencing NOTCH signaling causes growth arrest in both breast cancer stem cells and breast cancer cells. Br J Cancer 109: 2587–2596.
Suman S, Das TP, Moselhy J, Pal D, Kolluru V, Alatassi H, Ankem MK, Damodaran C (2016) Oral administration of withaferin A inhibits carcinogenesis of prostate in TRAMP model. Oncotarget 7: 53751–53761.
Suman S, Das TP, Reddy R, Nyakeriga AM, Luevano JE, Konwar D, Pahari P, Damodaran C (2014a) The pro-apoptotic role of autophagy in breast cancer. Br J Cancer 111: 309–317.
Suman S, Kurisetty V, Das TP, Vadodkar A, Ramos G, Lakshmanaswamy R, Damodaran C (2014b) Activation of AKT signaling promotes epithelial-mesenchymal transition and tumor growth in colorectal cancer cells. Mol Carcinog 53 (Suppl 1): E151–E160.
Szliszka E, Czuba ZP, Sedek L, Paradysz A, Krol W (2011) Enhanced TRAIL-mediated apoptosis in prostate cancer cells by the bioactive compounds neobavaisoflavone and psoralidin isolated from Psoralea corylifolia. Pharmacol Rep 63: 139–148.
Takenaka S, Oldiges H, Konig H, Hochrainer D, Oberdorster G (1983) Carcinogenicity of cadmium chloride aerosols in W rats. J Natl Cancer Inst 70: 367–373.
Turk B, Turk V (2009) Lysosomes as ‘suicide bags’ in cell death: myth or reality? J Biol Chem 284: 21783–21787.
U.S. Cancer Statistics Working Group (2016) United States Cancer Statistics: 1999–2013 Incidence and Mortality Web-based Report. U.S. Department of Health and Human Services, Centers for Disease Control and Prevention and National Cancer Institute: Atlanta, GA, USA. https://nccd.cdc.gov/uscs/.
Urani C, Melchioretto P, Fabbri M, Bowe G, Maserati E, Gribaldo L (2014) Cadmium Impairs p53 Activity in HepG2 Cells. ISRN Toxicol 2014: 976428.
Vemana G, Hamilton RJ, Andriole GL, Freedland SJ (2014) Chemoprevention of prostate cancer. Annu Rev Med 65: 111–123.
Waalkes MP (2003) Cadmium carcinogenesis. Mutat Res 533: 107–120.
Waalkes MP, Coogan TP, Barter RA (1992) Toxicological principles of metal carcinogenesis with special emphasis on cadmium. Crit Rev Toxicol 22: 175–201.
Wang J, Eltoum IE, Lamartiniere CA (2007) Genistein chemoprevention of prostate cancer in TRAMP mice. J Carcinog 6: 3.
Wu J, Dang Y, Su W, Liu C, Ma H, Shan Y, Pei Y, Wan B, Guo J, Yu L (2006) Molecular cloning and characterization of rat LC3A and LC3B—two novel markers of autophagosome. Biochem Biophys Res Commun 339: 437–442.
Xiao G, Li G, Chen L, Zhang Z, Yin JJ, Wu T, Cheng Z, Wei X, Wang Z (2010) Isolation of antioxidants from Psoralea corylifolia fruits using high-speed counter-current chromatography guided by thin layer chromatography-antioxidant autographic assay. J Chromatogr A 1217: 5470–5476.
Xie J, Shaikh ZA (2006) Cadmium-induced apoptosis in rat kidney epithelial cells involves decrease in nuclear factor-kappa B activity. Toxicol Sci 91: 299–308.
Yang Z, Yang S, Qian SY, Hong JS, Kadiiska MB, Tennant RW, Waalkes MP, Liu J (2007) Cadmium-induced toxicity in rat primary mid-brain neuroglia cultures: role of oxidative stress from microglia. Toxicol Sci 98: 488–494.
Acknowledgements
The work was supported by SOM collaborative Matching Grant to JF and CD.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Pal, D., Suman, S., Kolluru, V. et al. Inhibition of autophagy prevents cadmium-induced prostate carcinogenesis. Br J Cancer 117, 56–64 (2017). https://doi.org/10.1038/bjc.2017.143
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2017.143
Keywords
This article is cited by
-
Psoralidin inhibits osteosarcoma growth and metastasis by downregulating ITGB1 expression via the FAK and PI3K/Akt signaling pathways
Chinese Medicine (2023)
-
Unboxing the molecular modalities of mutagens in cancer
Environmental Science and Pollution Research (2022)
-
Chronic exposure to cadmium induces a malignant transformation of benign prostate epithelial cells
Oncogenesis (2020)
-
Toxicological effects of toxic metals (cadmium and mercury) on blood and the thyroid gland and pharmacological intervention by vitamin C in rabbits
Environmental Science and Pollution Research (2019)

{kind=link}