
Abstract
Background:
The primary objective was to determine whether topically administered Caphosol, rinsed orally four times daily at the initiation of conditioning, reduces the duration of severe oral mucositis (OM) compared with placebo among children and adolescents undergoing haematopoietic cell transplantation (HCT).
Methods:
This was a Children’s Oncology Group multicentre randomised double-blinded placebo-controlled clinical trial. Patients between the ages of 4 and 21 years who were scheduled to undergo myeloablative HCT for any indication were randomised to Caphosol or placebo saline rinses four times daily from initiation of conditioning through day +20. Subjects were assessed daily for OM using the World Health Organisation (WHO) Oral Toxicity Scale, Mouth Pain Categorical Scale (0–10) and the Oral Mucositis Daily Questionnaire (OMDQ). The primary end point was duration of severe OM (WHO ⩾3).
Results:
The study enrolled 220 participants with a median age of 13.7 years (range 4.0–21.9); 163 (74%) received allogeneic HCT. The mean (±s.d.) duration of severe OM was not reduced among Caphosol (4.5±5.0 days) vs placebo (4.5±4.8; P=0.99) recipients. The incidence of severe OM in the Caphosol and placebo arms was 63% (57 out of 91) and 68% (62 out of 91), respectively (P=0.44). There were no significant differences in any of the secondary end points between the groups.
Conclusions:
Caphosol did not reduce severe OM when compared with placebo among children and adolescents undergoing myeloablative HCT. Studies to identify effective interventions for OM are needed in this population.
Similar content being viewed by others
Main
Oral mucositis (OM) is a painful and debilitating complication that affects over 70% of children and adolescents undergoing haematopoietic cell transplantation (HCT) (Scully et al, 2006; Tomlinson et al, 2007; Vagliano et al, 2011). Clinical features of OM include diffuse mucosal erythema and ulcerations with accompanying mouth and throat pain, resulting in limited oral intake and suffering that, when severe, frequently requires opioid analgesics and total parenteral nutritional support (Sonis, 2004). In addition, the presence of severe OM has been associated with an increased risk of bacteraemia as well as prolonged length of hospital stay and greater total associated hospitalisation costs (Bochud et al, 1994; Sonis et al, 2001; Elting et al, 2003). Children consistently report OM as the most distressing aspect of the HCT experience (Ljungman et al, 1999; Bellm et al, 2000; Cheng, 2009). Although palifermin, a keratinocyte growth factor, is approved by the US Food and Drug Administration for prevention of OM in adults with haematologic malignancies undergoing HCT, it is expensive, and there is limited safety and efficacy data in children (Spielberger et al, 2004; Elting et al, 2007; Srinivasan et al, 2012; Nooka et al, 2014; Vitale et al, 2014). Prevention and optimal management of OM remains an important unmet need in pediatric HCT (Sung et al, 2015).
Caphosol (EUSA Pharma, Hemel Hempstead, UK) is a super-saturated calcium phosphate electrolyte solution that is approved by the FDA as a device to be used as an adjunct to standard oral care in patients with chemotherapy-associated OM (http://www.accessdata.fda.gov/cdrh_docs/pdf3/k030802.pdf). The mechanism of action of Caphosol remains to be elucidated, but it is thought that the high ionic solution permeates the oral mucosa and actives different signalling pathways, resulting in apoptosis modulation, downregulation of mediators of pain and inflammation, and activation of wound healing and epithelial proliferation (An et al, 1998). A single-centre phase III randomised controlled trial that evaluated the efficacy of Caphosol in preventing OM in adult patients undergoing HCT demonstrated statistically significant decreases in days of mucositis, pain and morphine use, as well as total dose of morphine (Papas et al, 2003). In all clinical reports, Caphosol has been consistently well tolerated with no reported side effects or toxicities (Quinn, 2013). However, the efficacy of Caphosol has not been replicated in other blinded and randomised studies and, in particular, definitive studies in pediatric patients have been few (Raphael et al, 2014). The primary objective of this Children’s Oncology Group (COG) multicentre trial was to determine whether topically administered Caphosol, rinsed orally four times daily at the initiation of conditioning, reduces the duration of severe OM compared with placebo saline solution among children and adolescents undergoing myeloablative HCT.
Materials and methods
Study design
This was a phase III international multicentre randomised double-blinded placebo-controlled clinical trial. This study was registered (NCT01305200) and reporting follows the CONSORT criteria (Rennie, 2001). The study was activated in April 2011 and was closed to enrolment in June 2014. The study was approved by the National Cancer Institute Central institutional review board (IRB) and the IRB of each participating centre. All patients and their parents or legal guardians provided written informed consent or assent as appropriate.
Patients
Patients between the ages of 4 and 21 years who were scheduled to undergo myeloablative autologous or allogeneic HCT for any indication including malignant and nonmalignant conditions were eligible for study participation (Bacigalupo et al, 2009). Permitted graft sources included bone marrow (BM), umbilical cord blood (UCB), or cytokine-mobilised peripheral blood stem cells (PBSCs). Eligible allogeneic donors were any human leukocyte antigen (HLA)-matched donor or partially matched family members (mismatched for a single HLA locus at A, B, C, or DR), unrelated BM, PBSCs, or UCB (at least 4 of 6 loci match at A, B, and DR). Eligible patients could not have received palifermin within 30 days before enrolment, and could not have been previously treated with Caphosol.
Study interventions
Caphosol was supplied as two separately packaged aqueous solutions, a 15 ml phosphate solution (Caphosol A) and a 15 ml calcium solution (Caphosol B), that were mixed to form a pH neutral supersaturated solution at the time of use. Caphosol was provided and distributed directly to the study centres by Jazz Pharmaceuticals (Dublin, Ireland). Placebo was sterile 0.9% sodium chloride injection solution that was supplied by each site. Saline stored in plastic bottles (e.g., single dose preservative free 0.9% sodium chloride injection used for drug reconstitution) was used instead of saline stored in polyvinyl chloride bags as the bags can impart an unpleasant smell and therefore could potentially compromise blinding.
Randomisation, allocation concealment, and blinding
Patients were randomised at enrolment to receive either Caphosol or placebo in a 1 : 1 ratio. Randomisation was stratified by type of graft (autologous vs allogeneic) and conditioning regimen (total body irradiation (TBI) or melphalan, vs neither TBI nor melphalan) and was performed in blocks of four. The allocation sequence was computer generated by the COG statistical office and allocation remained concealed to all health-care providers, children and their families, and outcome assessors.
To maintain blinding of the patients, health-care providers, and outcome assessors, the following series of procedures were performed. Each institution had two designated unblinded site pharmacists who were not directly involved in the care of study patients. They were specifically instructed to not share allocation with anyone at the site (provider or patient). For each randomised patient, the unblinded pharmacist was responsible for dispensing of the randomised intervention as follows. Upon enrolment of each participant, the unblinded pharmacist contacted the COG Research Coordinator by telephone, who verbally communicated the treatment assignment to the pharmacist. The pharmacist was asked to verbally repeat the treatment assignment for confirmation. The treatment assignment was also emailed to the pharmacist. The pharmacist then dispensed Caphosol or placebo as specified below.
Study rinse preparation and administration
Caphosol components (Caphosol A and Caphosol B, 2 × 15 ml) or placebo (2 × 15 ml of 0.9% sodium chloride solution) were transferred by the pharmacist into two identical oral syringes (marked ‘Syringe A’ and ‘Syringe B’) up to 4 days before use and delivered to the patient’s ward where they were accessible by the patient’s bedside nurse. At the time of planned administration, bedside nurses were instructed to mix the two syringes (Syringe A and Syringe B) into a cup and then to have the participant perform two rinses within 15 min of mixing. Nurses provided half of the mixed solution (15 ml) for the subject to rinse their mouth thoroughly and gargle for 1 min and spit. The same procedure was repeated with the remaining 15 ml of solution, for a total rinse time of 2 min. For younger children with small mouths who were unable to tolerate the full 15 ml of mixed solution, the volume of the mixture could be reduced to 5 to 10 ml, as tolerated. Subjects were instructed not to swallow the solution, and to avoid eating or drinking for 15 min after use.
Study therapy administration was initiated on the first day of conditioning and continued daily until day +20 (20 days following the date of stem cell infusion) or hospital discharge, whichever occurred first. Subjects rinsed four times per day (two rinses per episode) at approximately evenly spaced intervals. Subjects who developed World Health Organisation (WHO) Grade ⩾3 OM were permitted to request up to two additional study rinses per day, for a total of six rinses. In the event of a missed dose, every effort was made to make up the missed dose before bed, regardless of dosing interval, but individual doses could not be combined or ‘doubled-up’.
Outcomes
The primary outcome was the WHO Oral Toxicity grading scale (WHO, 1979). The WHO scale is the most commonly used scale in mucositis clinical trials and it consists of subjective, objective, and functional components, resulting in a score that ranges from 0 to 4 (0=no mucositis; 1=oral soreness and erythema; 2=ulcers and erythema, can swallow solid food; 3=ulcers and extensive erythema, cannot swallow solid food and can tolerate soft/liquid diet only; and 4=ulcers and extensive erythema, alimentation impossible). Subjects were queried regarding their ability to eat solids, liquids only, or nothing by mouth during the prior 24 h period. If the answer was ‘liquids’ or ‘nothing by mouth’, it was determined whether the limitation was due to OM-associated symptoms or due to other reasons such as nausea or anorexia. If diet limitations were not attributed to OM, then subjects were asked what they would otherwise be able to tolerate, and this answer was used to generate the WHO score.
In order to apply the WHO score, the assessor had to determine the presence or absence of erythema and ulcers. The assessor was instructed to examine the oral cavity in a site-directed manner and erythema or ulceration were documented for each of the following sites: lower labial mucosa, upper labial mucosa, right buccal mucosa, left buccal mucosa, right ventrolateral tongue, left ventrolateral tongue, floor of mouth, and soft palate. The WHO score was determined based on the diet answer, presence or absence of mouth pain, and clinical exam findings.
Secondary outcomes included severity of OM according to a Mouth Pain Categorical Rating Scale and Oral Mucositis Daily Questionnaire (OMDQ); incidence, total dose, and duration of opioid analgesic use (morphine equivalents); incidence and duration of total parenteral nutrition (TPN) administration; and the incidence of fever and neutropenia and invasive bacterial infections. Mouth pain experienced in the last 24 h period was rated from 0 to 10, with 0 being no pain and 10 being the worst pain imaginable. A modified version of the Oral Mucositis Daily Questionnaire (with diarrhoea-related questions removed), previously validated in children, was completed daily (Tomlinson et al, 2011; Manji et al, 2012). The OMDQ consists of a 0–4 Likert scale for each of six subscales consisting of mouth and throat pain and impact of mouth and throat soreness on sleeping, swallowing, drinking, eating, and talking. A summary score is not available for the OMDQ.
The incidence and duration of TPN, fever and neutropenia, invasive bacterial infections, and opioid analgesic drug, dose, and duration were abstracted from patient medical records from initiation of study rinses through day +20 or hospital discharge, death, or withdrawal from study, whichever occurred first. Fever and neutropenia was defined as a single oral temperature of >38.3 °C or a sustained temperature of ⩾38 °C for more than 1 h occurring during neutropenia (absolute neutrophil count <500 per μl or between 500 and 1000 per μl and falling). An invasive bacterial infection was defined as one or more cultures positive for a bacterial pathogen obtained from a blood culture. All opioids were converted to morphine equivalents using commonly accepted standards for comparisons of dose (Kamdar, 2008; Swarm et al, 2010; Chou et al, 2014; http://www.micromedexsolutions.com). Excluded from these calculations were nonopioids reported (n=3), when sites reported ‘dose not available/not correct’ (n=2), and missing duration (n=1).
Mucositis ascertainment
Subjects were assessed daily for OM by trained study staff during the observation period. The trained assessors completed the WHO scale, pain scale, and OMDQ by direct inquiry with the participant. Assessors consisted of physicians, nurses, and other clinical staff. Standardised OM assessment training was conducted in person at COG meetings as well as via live webinars. Only qualified and trained individuals were permitted to conduct OM assessments. Daily assessments (including weekends and holidays) were conducted starting from day −1 of HCT and continued until: (1) refusal of further participation for OM assessment; (2) day +20; or (3) discharge home, whichever occurred first. Even if the oral rinses were not performed, daily OM assessments were continued. In the event that oral assessments could not be performed on a given day (e.g., patient intubated in intensive care), they were resumed as soon as possible. Consistency in the assessor for each participant was strongly encouraged when possible.
Toxicity assessment
Subjects were assessed daily for study rinse-related toxicities using the National Cancer Institute’s Common Terminology Criteria for Adverse Events v3.0. The following adverse events were reported: ⩾Grade 3 allergic reaction, ⩾Grade 3 anaphylaxis, ⩾Grade 3 oral pain, ⩾Grade 2 dysgeusia, and ⩾Grade 3 infection with microbiologically documented positive blood cultures.
Statistical analysis
The primary study end point was duration of severe OM (defined as WHO score of 3 or 4) assessed daily from day −1 through day +20 (22 days). Patients with less than half of daily WHO assessments (<11 assessments) were deemed not evaluable for OM outcomes and excluded from all OM outcome analyses. However, all participants were included in the analyses of non-OM secondary end points and toxicities. The planned sample size of 200 evaluable patients (100 per arm) assumed a mean duration of severe OM of 5 days in the placebo arm vs 3 days in the Caphosol arm (s.d. of 5 days) and was based on a two-sample t-test with at least 80% power at two-sided α of 0.05. Maximum study enrolment was set at 235 patients to account for ineligible and inevaluable enrolments.
For evaluable patients with missing daily WHO assessments, missing daily assessments at the end of the assessment period were imputed by the last observation carried forward; missing daily assessments at the start of the assessment period were imputed by the median of the observations from other patients in the same stratum; intermittent missing daily assessments in-between two nonmissing assessments were imputed by linear interpolation. As the WHO score imputed by linear interpolation may not take integer values 1 to 4, an imputed WHO score of ⩾2.5 was considered severe OM.
Area under the curve (AUC) of the OMDQ subscales and Pain Categorical Rating Scale from day −1 to day +20 was calculated for patients evaluable for OM outcomes. In AUC calculation, missing daily assessments were imputed the same way as missing WHO score imputation described above. The missing data imputation approach for AUC only affected the AUC value if there were missing data at the start or the end of the assessment period, as AUC in-between two nonmissing assessments was identical to the missing data imputation by linear interpolation.
Approach was intention to treat and all participants were analysed in the arm to which they were randomised. Randomised patients not evaluable for OM outcomes (as described above) or randomised patients without data for secondary end points were excluded in the respective analyses. Continuous measures were compared between the two arms by two-sample t-tests. Categorical measures were compared between the two arms by χ2 test. All tests were two sided with P-values of <0.05 considered statistically significant.
Results
Patient characteristics
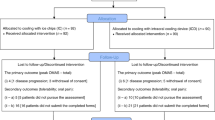
A total of 226 participants were identified; 4 were not eligible (mismatched donor, n=1; nonmyeloablative conditioning, n=2; incorrect short-form consent signed, n=1) and 2 were not randomised (sites were unable to reach the coordinating office because of time change difference or holidays). Thus, 220 eligible children and adolescents were randomised with a median age of 13.7 years (range 4–21.9 years; Table 1). Among the 220 randomised participants, 10 did not receive any protocol treatment (6 in Caphosol arm and 4 in placebo arm; Figure 1) because of subject refusal (n=5), consent withdrawal (n=3), or physician choice (n=2). The majority were being treated for malignant conditions and received primarily bone marrow or PBSCs grafts. Most participants underwent allogeneic HCT, with the majority receiving conditioning regimens that included TBI or melphalan.

Flow diagram of patient enrolment, allocation, and analysis. *Site unable to contact randomisation centre and subjects therefore never randomised before starting conditioning.
Study rinse and OM assessment compliance
Of the 220 (110 per arm) randomised subjects, 158 (72%, 79 per arm) received ⩾2 rinses per day on average (mean rinses per day=3.6; mean total rinses=95.5), and 62 (28%, 31 per arm) received <2 rinses per day on average (mean rinses per day=0.72; mean total rinses=20.2). With respect to age, subjects 4–12 years old completed a mean±s.d. of 2.5±1.5 rinses per day and subjects >12–21 years old completed a mean of 3.0±1.3 rinses per day. There was no difference in mean rises per day (placebo=3.6 (s.e.=0.05) vs treatment=3.6 (s.e.=0.05), P=0.70) or mean total rinses (placebo=96.0 (s.e.=2.0) vs treatment=95.0 (s.e.=2.0), P=0.72) between treatment arms.
There were 38 subjects (19 in each arm) who were inevaluable for the primary end point because of an insufficient number of completed WHO assessments (<11 days). All of the participants who were enrolled but did not receive any protocol treatment (n=10) were inevaluable because of the absence of any mucositis assessments. Of the 38 inevaluable subjects, 24 had no OM assessments. The 182 patients (182 out of 220, 83%) with ⩾11 daily WHO scores (mean=19.7, range 11–22) were therefore evaluable for primary and secondary end point analyses. Evaluable participants, when compared with nonevaluable participants, were not significantly different with respect to the patient characteristics variables reported in Table 1 (data not shown).
WHO Oral Toxicity Score end points
Table 2 and Figure 2 illustrate that with respect to the primary study end point, the mean±s.d. days of severe OM was not reduced among Caphosol (4.5±5.0 days) vs placebo (4.5±4.8 days; P=0.99) recipients. The mean treatment difference between study arms was 0 (95% confidence interval −1.4 to 1.4). When limiting the primary analysis to subjects with an average of ⩾2 daily rinses, there was no difference in mean±s.d. days of severe mucositis between the Caphosol (4.3±5.0 days) and placebo (4.2±4.7 days; P=0.88) arms. The incidence of severe OM (WHO ⩾3) in the Caphosol and placebo arms was 63% (57 out of 91) and 68% (62 out of 91), respectively (P=0.44).

Average daily mucoistis scores among those randomised to caphosol vs placebo. Average daily mucositis scores include nonimputed data.
Secondary end points
There were no statistically significant differences in the Mouth Pain Categorical Rating Scale, OMDQ subscales, incidence, duration or total dose of opioids, incidence and duration of TPN, incidence of fever and neutropenia, or incidence of invasive bacterial infection between the Caphosol and placebo arms (Tables 2 and 3 and Supplementary Table 1).
Adverse events
There were no toxicities that were attributed to study rinses. There were three deaths reported on treatment or within 1 month of the end of treatment, none of which were attributed to study rinses.
Discussion
In this international, multicentre randomised double-blinded trial, Caphosol did not reduce the duration of severe OM (WHO score ⩾3) compared with placebo in pediatric and adolescent patients undergoing myeloablative HCT. There were also no differences between the two arms in any of the study’s secondary end points. Consequently, this study does not support the routine use of Caphosol for prevention of OM in children and adolescents undergoing myeloablative HCT.
Our results are concordant with other recently reported prospective, randomised, placebo-controlled studies evaluating Caphosol for prevention of OM in HCT. Raphael et al (2014) reported a prospective multicentre double-blind randomised controlled trial evaluating interventional (rather than preventive) Caphosol vs 0.9% saline placebo in children (4–18 years old) undergoing chemotherapy alone or HCT. Study rinses were initiated following onset of OM, and all end points between the two groups were similar. Svanberg et al (2015) reported a randomised controlled open-abel trial of cryotherapy with or without Caphosol for prevention of OM in patients undergoing allogeneic HCT, with no differences in OM between the two groups.
As a topically applied agent, compliance with daily Caphosol rinses would be expected to be an important factor affecting treatment efficacy and effectiveness. Compliance with the prescribed study rinses was not described in any of the prior reports of Caphosol for OM prevention, and this is an important limitation of those publications. In our study, of the 220 randomised patients, 72% received ⩾2 rinses per day on average with no difference between the two study arms. This finding suggests that oral rinses that require multiple daily administrations may not be feasible in children and adolescents undergoing HCT as 28% were not able to maintain this intensity of oral rinses. Although this observation provides important insight into regimens likely to be successful in future studies, the sensitivity analysis among patients receiving on average two rinses per day also failed to show a benefit of Caphosol, further strengthening our conclusion of its lack of efficacy.
In the current study, rinses were generally well tolerated, although there were some anecdotal reports describing an unpleasant ‘salty’ taste. The study by Wasko-Grabowska et al (2011) described one subject (1 out of 32, 3.1%) who had ‘problems with the taste’ of Caphosol, but any potential impact on regimen compliance was not reported. The study by Svanberg et al (2015) reported one subject who stopped Caphosol rinses because of disagreeable taste. Of note, the study by Raphael et al (2014) was conducted in children aged 4 to 18 years randomised to NaCl 0.9% or Caphosol and reported that no patients dropped out because of intolerance/bad taste, although compliance was not described. In the current study, the reason for missed rinses was only collected when no daily rinses were completed, but not when less than four were completed; therefore, the true extent to which taste/tolerability (or other factors) affected compliance (in either study arm) is unknown.
This study had a number of strengths and potential advantages over prior Caphosol studies. This was a well-designed centrally administrated prospective international multicentre randomised placebo-controlled clinical trial with objectively measured end points. All participating study staff members were trained either in person or by webinar to ensure consistent study rinse administration, OM evaluation, data collection, and submission across sites and investigators. Furthermore, pediatric-specific considerations in how to administer the WHO Toxicity Scale are rarely considered but important as children undergoing cancer therapy do not eat or drink for many reasons other than mouth pain (Tomlinson et al, 2008). The study accrued more quickly than anticipated, highlighting the enthusiasm among investigators in finding a safe and effective intervention to limit the morbidity associated with OM.
However, our study should be interpreted in light of its weaknesses. Our study, likely many mucositis trials, was limited by missing data and nonevaluable subjects. In addition, the patient population was heterogeneous and supportive care was not standardised. However, these aspects should not have greatly affected our trial results as all HCT regimens were considered myeloablative and there are a paucity of approaches known to reduce OM in this patient population. Furthermore, although all patients underwent dental clearance per institutional guidelines before the start of the transplant conditioning regimen, we did not collect pretransplant dental evaluation or ongoing oral care, although we have no reason to believe oral health status or care would have differed between randomised groups. Finally, the number of final evaluable subjects was lower than the originally planned sample size, and thus our study was underpowered to statistically demonstrate the specified effect size. However, description of the difference and 95% CI around the primary end point illustrates a lack of difference between randomised groups.
In conclusion, Caphosol did not reduce severe OM when compared with placebo among children and adolescents undergoing myeloablative HCT. Studies to identify effective interventions for OM are needed in this population.
Change history
03 January 2017
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
An S, Goetzl EJ, Lee H (1998) Signaling mechanisms and molecular characteristics of G protein-coupled receptors for lysophosphatidic acid and sphingosine 1-phosphate. J Cell Biochem Suppl 30–31: 147–157.
Bacigalupo A, Ballen K, Rizzo D, Giralt S, Lazarus H, Ho V, Apperley J, Slavin S, Pasquini M, Sandmaier BM, Barrett J, Blaise D, Lowski R, Horowitz M (2009) Defining the intensity of conditioning regimens: working definitions. Biol Blood Marrow Transplant 15 (12): 1628–1633.
Bellm LA, Epstein JB, Rose-Ped A, Martin P, Fuchs HJ (2000) Patient reports of complications of bone marrow transplantation. Support Care Cancer 8 (1): 33–39.
Bochud PY, Calandra T, Francioli P (1994) Bacteremia due to viridans streptococci in neutropenic patients: a review. Am J Med 97 (3): 256–264.
Cheng KK (2009) Oral mucositis: a phenomenological study of pediatric patients' and their parents' perspectives and experiences. Support Care Cancer 17 (7): 829–837.
Chou R, Cruciani RA, Fiellin DA, Compton P, Farrar JT, Haigney MC, Inturrisi C, Knight JR, Otis-Green S, Marcus SM, Mehta D, Meyer MC, Portenoy R, Savage S, Strain E, Walsh S, Zeltzer L American Pain Society; Heart Rhythm Society (2014) Methadone safety: a clinical practice guideline from the American Pain Society and College on Problems of Drug Dependence, in collaboration with the Heart Rhythm Society. J Pain 15 (4): 321–337.
Elting LS, Cooksley C, Chambers M, Cantor SB, Manzullo E, Rubenstein EB (2003) The burdens of cancer therapy. Clinical and economic outcomes of chemotherapy-induced mucositis. Cancer 98 (7): 1531–1539.
Elting LS, Shih YC, Stiff PJ, Bensinger W, Cantor SB, Cooksley C, Spielberger R, Emmanoulides C (2007) Economic impact of palifermin on the costs of hospitalization for autologous hematopoietic stem-cell transplant: analysis of phase 3 trial results. Biol Blood Marrow Transplant 13 (7): 806–813.
Kamdar M (2008) Principles of Analgesic Use in the Treatment of Acute Pain and Cancer Pain Vol. 6th (edn) American Pain Society: Glenview, IL, USA.
Ljungman G, Gordh T, Sorensen S, Kreuger A (1999) Pain in paediatric oncology: interviews with children, adolescents and their parents. Acta Paediatr 88 (6): 623–630.
Manji A, Tomlinson D, Ethier MC, Gassas A, Maloney AM, Sung L (2012) Psychometric properties of the Oral Mucositis Daily Questionnaire for child self-report and importance of mucositis in children treated with chemotherapy. Support Care Cancer 20 (6): 1251–1258.
Nooka AK, Johnson HR, Kaufman JL, Flowers CR, Langston A, Steuer C, Graiser M, Ali Z, Shah NN, Rangaraju S, Nickleach D, Gao J, Lonial S, Waller EK (2014) Pharmacoeconomic analysis of palifermin to prevent mucositis among patients undergoing autologous hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 20 (6): 852–857.
Papas AS, Clark RE, Martuscelli G, O'Loughlin KT, Johansen E, Miller KB (2003) A prospective, randomized trial for the prevention of mucositis in patients undergoing hematopoietic stem cell transplantation. Bone Marrow Transplant 31 (8): 705–712.
Quinn B (2013) Efficacy of a supersaturated calcium phosphate oral rinse for the prevention and treatment of oral mucositis in patients receiving high-dose cancer therapy: a review of current data. Eur J Cancer Care (Engl) 22 (5): 564–579.
Raphael MF, den Boer AM, Kollen WJ, Mekelenkamp H, Abbink FC, Kaspers GJ, Zomer-Kooijker K, Molmans BH, Tissing WJ (2014) Caphosol, a therapeutic option in case of cancer therapy-induced oral mucositis in children?: Results from a prospective multicenter double blind randomized controlled trial. Support Care Cancer 22 (1): 3–6.
Rennie D (2001) CONSORT revised—improving the reporting of randomized trials. JAMA 285 (15): 2006–2007.
Scully C, Sonis S, Diz PD (2006) Oral mucositis. Oral Dis 12 (3): 229–241.
Sonis ST (2004) The pathobiology of mucositis. Nat Rev Cancer 4 (4): 277–284.
Sonis ST, Oster G, Fuchs H, Bellm L, Bradford WZ, Edelsberg J, Hayden V, Eilers J, Epstein JB, LeVeque FG, Miller C, Peterson DE, Schubert MM, Spijkervet FK, Horowitz M (2001) Oral mucositis and the clinical and economic outcomes of hematopoietic stem-cell transplantation. J Clin Oncol 19 (8): 2201–2205.
Spielberger R, Stiff P, Bensinger W, Gentile T, Weisdorf D, Kewalramani T, Shea T, Yanovich S, Hansen K, Noga S, McCarty J, LeMaistre CF, Sung EC, Blazar BR, Elhardt D, Chen MG, Emmanouilides C (2004) Palifermin for oral mucositis after intensive therapy for hematologic cancers. N Engl J Med 351 (25): 2590–2598.
Srinivasan A, Kasow KA, Cross S, Parrish M, Wang C, Srivastava DK, Cai X, Panetta JC, Leung W (2012) Phase I study of the tolerability and pharmacokinetics of palifermin in children undergoing allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 18 (8): 1309–1314.
Swarm R (2010) Clinical practice guidelines in oncology: adult cancer pain. J Natl Comprehensive Cancer Netw 8 (9): 1046–1086.
Sung L, Robinson P, Treister N, Baggott T, Gibson P, Tissing W, Wiernikowski J, Brinklow J, Dupuis LL (2015) Guideline for the prevention of oral and oropharyngeal mucositis in children receiving treatment for cancer or undergoing haematopoietic stem cell transplantation. BMJ Support Palliat Care e-pub ahead of print 27 March 2015 doi:10.1136/bmjspcare-2014-000804.
Svanberg A, Ohrn K, Birgegard G (2015) Caphosol((R)) mouthwash gives no additional protection against oral mucositis compared to cryotherapy alone in stem cell transplantation. A pilot study. Eur J Oncol Nurs 19 (1): 50–53.
Tomlinson D, Ethier MC, Judd P, Doyle J, Gassas A, Naqvi A, Sung L (2011) Reliability and construct validity of the oral mucositis daily questionnaire in children with cancer. Eur J Cancer 47 (3): 383–388.
Tomlinson D, Gibson F, Treister N, Baggott C, Judd P, Hendershot E, Maloney AM, Doyle J, Feldman B, Sung L (2008) Challenges of mucositis assessment in children: expert opinion. Eur J Oncol Nurs 12 (5): 469–475.
Tomlinson D, Judd P, Hendershot E, Maloney AM, Sung L (2007) Measurement of oral mucositis in children: a review of the literature. Support Care Cancer 15 (11): 1251–1258.
Vagliano L, Feraut C, Gobetto G, Trunfio A, Errico A, Campani V, Costazza G, Mega A, Matozzo V, Berni M, Alberani F, Banfi MM, Martinelli L, Munaron S, Orlando L, Lubiato L, Leanza S, Guerrato R, Rossetti A, Messina M, Barzetti L, Satta G, Dimonte V (2011) Incidence and severity of oral mucositis in patients undergoing haematopoietic SCT—results of a multicentre study. Bone Marrow Transplant 46 (5): 727–732.
Vitale KM, Violago L, Cofnas P, Bishop J, Jin Z, Bhatia M, Kung AL, George D, Garvin J, Satwani P (2014) Impact of palifermin on incidence of oral mucositis and healthcare utilization in children undergoing autologous hematopoietic stem cell transplantation for malignant diseases. Pediatr Transplant 18 (2): 211–216.
Wasko-Grabowska A, Rzepecki P, Oborska S, Barzal J, Gawronski K, Mlot B, Szczylik C (2011) Efficiency of supersaturated calcium phosphate mouth rinse treatment in patients receiving high-dose melphalan or BEAM prior to autologous blood stem cell transplantation: a single-center experience. Transplant Proc 43 (8): 3111–3113.
WHO (1979) World Health Organization Handbook for Reporting Results of Cancer Treatment. WHO: Geneva.
Acknowledgements
We thank the patients, families, and study staffs from all of the participating study sites in the United States, Canada, and Australia/New Zealand. We thank the Children’s Oncology Group Operations Center for coordinating the randomisation process. We thank Jazz Pharmaceuticals for supplying and shipping Caphosol to the study sites. This research was supported by the following grants from the National Cancer Institute, National Institutes of Health: The Children’s Oncology Group Chair’s Grant U10CA98543–08, NCTN Operations Center Grant U10CA180886, NCTN Statistics & Data Center U10CA180899, and Statistics & Data Center Grant U10CA098413.
Author contributions
Study design: Nathaniel Treister, Michael Nieder, Christina Baggott, Ellen Olson, Lu Chen, and Lillian Sung; data analysis: Nathaniel Treister, Michael Nieder, Christina Baggott, Ellen Olson, Lu Chen, Ha Dang, Mark Krailo, Amanda August, and Lillian Sung; manuscript drafting: Nathaniel Treister, Michael Nieder, Christina Baggott, Ellen Olson, Lu Chen, Ha Dang, Mark Krailo, Amanda August, and Lillian Sung.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Treister, N., Nieder, M., Baggott, C. et al. Caphosol for prevention of oral mucositis in pediatric myeloablative haematopoietic cell transplantation. Br J Cancer 116, 21–27 (2017). https://doi.org/10.1038/bjc.2016.380
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2016.380
Keywords
This article is cited by
-
Nursing care approaches to oral mucositis in pediatric oncology clinics: a cross-sectional study from Türkiye
Supportive Care in Cancer (2023)
-
Oral hygiene protocols reduce the severity and incidence of oral mucositis during antineoplastic treatment: a systematic review and meta-analysis of randomized and non-randomized clinical trials
Supportive Care in Cancer (2023)
-
Nattokinase crude extract enhances oral mucositis healing
BMC Oral Health (2021)
-
Treatment for Oral Mucositis—Current Options and an Update of Small Molecules Under Development
Current Treatment Options in Oncology (2021)
-
MASCC/ISOO clinical practice guidelines for the management of mucositis: sub-analysis of current interventions for the management of oral mucositis in pediatric cancer patients
Supportive Care in Cancer (2021)
