
Abstract
Background:
The binding of STAT3 and STAT5 to growth factor and cytokine receptors such as EGFR and IL-6 receptor gp130 is critical to their activation and ability to contribute to malignant transformation. Therefore, interfering with these biochemical processes could lead to the discovery of novel anticancer agents.
Methods:
Co-immunoprecipitation, western blotting, microscopy, DNA binding, invasion, and soft agar assays as well as a mouse model were used to investigate the mechanism by which the natural product Withacnistin (Wit) inhibits STAT 3/5 tyrosine phosphoryaltion and activation.
Results:
Wit blocks EGF- and IL-6-stimulated binding of STAT3 and STAT5 to EGFR and gp130. Wit inhibits EGF-, PDGF-, IL-6-, IFNβ-, and GM-CSF-stimulation of tyrosine phosphorylation of STAT3 and STAT5 but not of EGFR or PDGFR. The inhibition of P-STAT3 and P-STAT5 occurred rapidly, within minutes of Wit treatment and growth factor stimulation. Wit also inhibits STAT3 nuclear translocation, DNA binding, promoter transcriptional activation, and it suppresses the expression levels of STAT3 target genes such as Bcl-xL and Mcl-1. Finally, Wit induces apoptosis, inhibits anchorage-dependent and -independent growth and invasion, and causes breast tumour regression in an ErbB2-driven transgenic mouse model.
Conclusions:
These data warrant further development of Wit as a novel anticancer drug for targeting tumours that harbour hyperactivated STAT3 and STAT5.
Similar content being viewed by others

Main
Signal transducers and activators of transcription (STAT) proteins are activated in response to various cytokines and growth factors (GFs) to mediate specific cellular responses. The activation of STAT proteins relies on their binding at the plasma membrane to GF and cytokine receptors and subsequent tyrosine phosphorylation, either directly by the receptor tyrosine kinases (RTKs) themselves (i.e., EGFR) or non-RTKs (i.e., Src or JAK) that are associated with cytokine receptors such as gp130 (Darnell, 2002; Yu et al, 2009). Upon phosphorylation, STATs dimerise via reciprocal SH2-domain/phosphotyrosine interactions, localise to the nucleus, bind to STAT-specific DNA response elements, and induce transcription of their target genes. The biochemical roles of the eight known STAT proteins (STATs 1A, 1B, 2, 3, 4, 5A, 5B, and 6) are diverse, ranging from development and cell differentiation and proliferation to regulation of apoptosis and inflammation and immune response (Darnell, 1997; Takeda et al, 1997; Yu and Jove, 2004).
Among the eight isoforms, STAT3 and STAT5 are found to be persistently phosphorylated and hyperactivated in many human cancers including lung, breast, colon, bladder, and other solid tumours, as well as haematological malignancies (Takeda et al, 1997; Sinibaldi et al, 2000; Yu and Jove, 2004). The constitutive activation of STAT3 and STAT5 has been shown to have vital roles in tumour proliferation, survival, and angiogenesis, as well as the immune response of tumours (Shao et al, 2003; Wang et al, 2004; Malin et al, 2010). The hyperactivation can occur through upregulation of RTKs (e.g., EGFR), cytokine receptors (e.g., gp130), mutation of non-RTK (e.g., JAK), or gene translocation resulting in abnormal fusion proteins (e.g., BCR-Abl) (Darnell, 2002; Coppo et al, 2003; Shao et al, 2003; Yu et al, 2009). In addition to being constitutively activated, STAT3 and STAT5 are required for malignant transformation of many human tumour types. For example, suppression of STAT3 by either a dominant-negative form (STAT3-β) or by siRNA induces apoptosis and blocks proliferation, invasion, angiogenesis, and/or tumour growth in vivo (Maritano et al, 2004; Dai et al, 2011). Similarly, inhibition of STAT5 with pimodize decreases survival, induces apoptosis, and suppresses cell proliferation of chronic myelogenous leukaemia cells (Nelson et al, 2011). The prevalent hyperactivation of STAT3 and its requirement for malignant transformation led to the development of many inhibitors that are being investigated preclinically (Page et al, 2011; Debnath et al, 2012). These include compounds that inhibit activators of STAT3, such as JAK2, and phosphotyrosine peptide mimics that disrupt STAT3–STAT3 dimerisation (Chen et al, 2007; Zhang et al, 2010; Mandal et al, 2011; Zhang et al, 2012, 2013). In contrast, only few STAT5 inhibitors have been developed (Bar-Natan et al, 2012; Page et al, 2012).
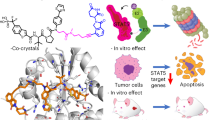
Our efforts to identify STAT3 pathway inhibitors resulted in the discovery of the natural product Withacnistin (Wit) that inhibits tyrosine phosphorylation of STAT3 without inhibiting the phosphorylation of JAK2, Src, Akt, Erk, or JNK (Sun et al, 2005; Sun et al, 2008). Wit is a member of the withanolide family found in such plants as Withania somnifera and Acnistus arborescens that have been used for centuries as remedies for several ailments including cancer (Groijourdy, 1864; F. Haussler, 1914; Kupchan et al, 1969; Deepak Mundkinajeddu et al, 2014). Withacnistin was identified by Kupchan et al (1969) from the leaves of Acnistus arborescens (L.) Schlecht (Solanaceae) that were collected in Costa Rica. In 2005, using a 96-well cytoblot to screen the NCI diversity set for compounds that decrease the levels of STAT3 phosphorylation in MDA-MB-468 breast cancer cells, we identified Wit as a potent hit (Sun et al, 2005; Sun et al, 2008). However, at present the mechanism by which Wit inhibits STAT3 tyrosine phosphorylation has not been investigated. Furthermore, it is also not known whether Wit inhibits tyrosine phosphorylation of STAT5, another oncogenic STAT protein. In this manuscript, we show that Wit rapidly inhibits not only basal levels of constitutively activated STAT3 but also inhibits growth factor- (e.g., EGF and PDGF) and cytokine- (e.g., IL-6, IFN-β and GM-CSF) stimulated STAT3 and STAT5 tyrosine phosphorylation. We have also determined that the mechanism by which this occurs is through inhibition of GF- and cytokine-stimulated STAT3/ and STAT5/receptor binding thus blocking their recruitment to the plasma membrane. We have also shown that the inhibition by Wit of STAT/receptor binding resulted in decreased tyrosine phosphorylation and subsequent inhibition of nuclear translocation, DNA binding, transcription, anchorage-independent growth, and invasion. Finally, we show in an ErbB2-driven transgenic mouse model of mammary oncogenesis that Wit treatment induces tumour regression.
Materials and Methods
Cell lines and reagents
All cell lines were obtained from ATCC (American Type Culture Collection, Manassas, VA, USA). These cell lines have not been authenticated. Human breast carcinoma MDA-MB-468, human lung cancer A549, and murine fibroblast NIH-3T3 cells were maintained in Dulbecco’s modified Eagle’s medium (Life Technologies Corporation, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS), 10 mM sodium pyruvate, 25 mM HEPES, pH 7.5, 1000 U ml−1 penicillin, and 1000 μg ml−1 streptomycin. Human multiple myeloma U266, RPMI 8226, and human erythroleukemia TF-1 cells were maintained in RPMI 1640 Medium (Life Technologies Corporation) supplemented with 15% (for U266) or 10% (TF-1) FBS, 10 mM sodium pyruvate, 25 mM HEPES, pH 7.5, 1000 U ml−1 penicillin, and 1000 μg streptomycin; in addition, TF-1 medium was supplemented with 2 ng ml−1 GM-CSF (Sigma-Aldrich, St Louis, MO, USA).
Primary antibodies against pY705-STAT3, pY694-STAT5, STAT5, EGFR, pY1068-EGFR, and Bcl-xl were purchased from Cell Signaling Technology (Danvers, MA, USA). Primary antibodies against STAT3 (C-20), Mcl-1, and gp130 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Withacnistin
Original powder stocks of Withacnistin were obtained from the National Cancer Institute Developmental Therapeutics Program. Subsequent Withacnistin stocks were then obtained from Dr Giselle Tamayo of the Instituto Nacional de Biodiversidad in Costa Rica. Withacnistin was dissolved in DMSO for use in these experiments. The purity of withacnistin used can be found in (Sun et al, 2008) and Supplementary Materials at: http://www.nature.com/onc/journal/v27/n9/suppinfo/1211028s1.html?url=/onc/journal/v27/n9/full/1211028a.html
Growth factor and cytokine stimulation
For serum starvation of MDA-MB-468 and U266, cells were incubated in the medium specified without supplemental FBS. For TF-1 starvation, medium contained neither FBS nor GM-CSF. For NIH-3T3 cells, the starvation medium consisted of the medium as described above, replacing 10% FBS with 0.5% FBS. After starvation for 24–48 h, cells were then pretreated with desired concentrations of Wit in serum-free medium containing 1 mg ml−1 bovine serum albumin (BSA) for the time indicated in the figure legends, then stimulated for 10 or 30 min with either 100 ng ml−1 EGF (Millipore, Billerica, MA, USA) for MDA-MB-468, 10 ng ml−1 PDGF-BB (Life Technologies Corporation) for NIH-3T3, 10 ng ml−1 IL-6 (Millipore) or 100 U ml−1 IFN-β (R&D Systems, Minneapolis, MN, USA) for U266, or 2 ng ml−1 GM-CSF for TF-1 cells. The reaction was stopped by the addition of ice-cold phosphate-buffered saline (PBS), pH 7.5.
Western blots
Cells were harvested and lysed for 30 min on ice with occasional vortexing in 150 mM Hepes, pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.5% NP-40, 10% glycerol, 5 mM NaF, 1 mM DTT, 1 mM phenylmethylsulfonyl fluoride (PMSF), 2 mM Na3VO4, and 5 μg ml−1 leupeptin. Proteins readings were done using the Bradford protein assay, and equal amounts of protein for each sample were loaded into the wells of SDS–PAGE gels. After separation, proteins were transferred to nitrocellulose and western blots were performed as we did previously (Zhang et al, 2013).
Immunoprecipitation
MDA-MB-468 cells, U266 cells, and RPMI 8226 cells were plated and grown to about 70% confluency then starved as described above for 24 h. After starvation, cells were pretreated for 5 min with 10 μ M Wit or with DMSO vehicle control and then stimulated for 30 min with 100 ng ml−1 EGF or 100 ng ml−1 IL-6, or with PBS containing 1% BSA (EGF vehicle). Reaction was halted, as above, with ice-cold PBS and cells were lysed on the plates in a small volume of 10 mM Tris, pH 7.5, 150 mM NaCl, 10% glycerol, 5 mM EDTA, 1% Triton-X-100, 0.1% SDS, 10 μg ml−1 aprotinin, 10 μg ml−1 soybean trypsin inhibitor, 25 μg ml−1 leupeptin, 2 mM PMSF, and 2 mM sodium orthovanadate for 30 min on ice. Cells were scraped into microcentrifuge vials and spun at 13 000 g to clear the lysates. Protein readings were done using the Bradford protein assay, then equal amounts of protein were immunoprecipitated as we described previously (Zhang et al, 2013).
STAT3 nuclear accumulation
MDA-MB-468 cells were plated at 4000 cells per well in eight-well chamber slides. The cells were treated the following day with vehicle or Wit for 30 min at different concentrations. Cells were starved and stimulated as described above. Cells were fixed using cold methanol for 15 min, washed with PBS three times, and permeabilised using 0.25% Triton-X-100 for 15 min. Specimens were then blocked in 1% BSA for 30 min and incubated with indicated antibodies at 1 : 50 dilution at 4 °C overnight. Subsequently, cells were rinsed 4–5 times in PBS, incubated with Alexa Fluor secondary antibody (Life Technologies, Long Island, NY, USA) for 1 h at room temperature in the dark. Specimens were then washed five times with PBS, slides mounted with VECTASHIELD mounting medium containing 4',6-diamidino-2-phenylindole (DAPI) (Vector Laboratories Inc., Burlingame, CA, USA), and examined immediately under a Zeiss Upright Fluorescence Microscope (Zeiss, Thornwood, NY, USA).
Nuclear extract preparation and STAT3 filter plate assay
Nuclear extract preparation was carried out as previously described (Zhang et al, 2010).The STAT3–DNA-binding filter plate assay was performed following the manual of the filter plate assay kit (Signosis, Sunnyvale, CA, USA), as described previously (Zhang et al, 2013). The TF-Binding buffer was mixed with the STAT3 probe (biotin-labelled STAT3–DNA-binding sequence) and nuclear extract and incubated at 16 °C for 30 min to form the STAT3–DNA complex. The STAT3–DNA complex was then separated from free probe by using a filter plate. After several steps of binding and washing, bound STAT3 probe is retained on the filter and the free DNA probe is removed. The bound pre-labelled STAT3 probe was then eluted from the filter plate by centrifugation with elution buffer. Eluted probes were then hybridised into 96-well hybridisation plates for quantitative analysis. The captured STAT3 probe was detected by reaction with streptavidin-HRP. The chemiluminescence of each well was read using 2104 EnVisionR Multilabel Reader (Perkin Elmer, Waltham, MA, USA) within 5 min after mixture with substrates.
STAT3 transcriptional activity
MDA-MB-468 cells were plated into 12-well plates at a density of 4 × 105 cells per well. The cells were transiently transfected with pLucTKS3/β-gal or pLucSRE/β-gal, and then were treated with vehicle and Wit for 48 h. Then cytosolic extracts of equal total protein were prepared from Wit-treated or -untreated cells and analysed for luciferase activity using a TD-20/20 luminometer (TurnerDesigns, Sunnyvale, CA, USA) described by us previously (Zhang et al, 2013).
Anchorage-independent growth by soft agar assay
Soft agar colony formation assays were performed in 12-well plate as described previously (Zhang et al, 2013). Briefly, cells were seeded at 2000 cells per well in regular growth media containing 0.3% agar (Sigma, St Louis, MO, USA) and Wit was added the following day. Colonies were allowed to grow for 3–4 weeks, and quantified by staining with 1 mg ml−1 MTT (Sigma) overnight.
Invasion assay
Invasion assay was performed in BD BioCoat Matrigel Invasion Chamber (BD Biosciences, San Jose, CA, USA) in 24-well plates. MDA-MB-468 cells were seeded at 25 000 cells per insert in the top chamber over the Matrigel. The bottom chamber contained 20% FBS as the chemoattractant. Vehicle and Wit were added the following day. The cells were incubated for 48 h, after which the cells in the top chamber were carefully removed and the filter membranes containing the invaded cells on the outside of the filter were fixed with methanol, stained with crystal violet, and photographed.
TUNEL assay
MDA-MB-468 cells were treated with increasing concentrations of Wit for either 24 or 48 h. After treatment, cells were harvested by trypsinisation and counted using the Trypan blue exclusion assay. Cells (75 000) were spun onto slides using a Cytospin centrifuge. After fixing cells to the slides with 4% paraformaldehyde in PBS (pH 7.5) for 1 h at room temperature, cells were labelled for apoptotic DNA strand breaks by TUNEL (Terminal deoxynucleotidyl transferase dUTP Nick-End Labelling) reaction using an in situ cell death detection kit, fluorescein (Roche Applied Science, Indianapolis, IN, USA), according to the manufacturer’s instructions. Slides were mounted in Vectashield mounting medium (Vector Laboratories) containing DAPI to counterstain DNA. Fluorescein-labelled DNA strand breaks (TUNEL-positive cells) were then visualised using a fluorescence microscope (Leica Microsystems Inc., Bannockburn, IL, USA), and pictures were taken with a digital camera (Diagnostic Instruments Inc., Sterling Heights, MI, USA). TUNEL-positive nuclei were counted and compared with DAPI-stained nuclei to determine the percentage induction of apoptosis by 10 μ M Wit compared with control-treated cells.
Antitumour activity in the ErbB2 mouse tumour model
MMTV/Neu transgenic mice (FVB/N-Tg(MMTVneu)202 Mul/J) were purchased from Jackson Laboratory (Bar Harbor, ME, USA) and bred to produce multiple litters for colony maintenance. Female mice were palpated once each week for mammary gland tumour nodules as described by us previously (Balasis et al, 2011). At the time of tumour onset, orthogonal measurements were taken 2–3 times per week and the tumour volume calculated using the formula: V=(a2b)/2, where ‘a’ and ‘b’ are defined as the smaller and larger measurements, respectively. Drug treatment was initiated when the tumours grew to about 1200–2400 mm3 and treatment lasted up to 14 days. Wit was administrated to ErbB2 tumour-bearing mice by i.p. injections at a dosage of 20 mpk per day. There was no evidence of gross toxicity in the drug-treated animals as measured by weight loss. All methods involving mice were approved by the Institutional Animal Care and Use Committee of the University of South Florida.
Results
Withacnistin (Wit) inhibits EGF- and IL-6-stimulated STAT3 and STAT5 recruitment to EGFR and gp130 receptors
Previously, we showed that Wit inhibits the constitutive levels of tyrosine-phosphorylated STAT3 in human tumours where STAT3 is hyperactive. However, the mechanism by which Wit reduces P-Tyr-STAT3 levels is not known. Furthermore, whether Wit also interferes with signalling of STAT5, another STAT protein involved in oncogenesis is not known. To address these important questions, first we determined whether Wit could interfere with the ability of growth factors such as EGF to stimulate the tyrosine phosphorylation of STAT3 and STAT5. To this end, we pretreated serum-starved MDA-MB-468 breast cancer cells with Wit for increasing lengths of time before EGF stimulation, and processed the cells for western blotting as described under Materials and Methods. Figure 1A shows that in the absence of Wit, EGF stimulated the tyrosine phosphorylation of both STAT3 and STAT5. Pretreatment with Wit for as little as 2–10 min inhibited EGF-stimulated STAT3 tyrosine phosphorylation partially, and complete inhibition occurred within 15–30 min. Figure 1A also shows that Wit pretreatment was able to inhibit EGF-stimulated STAT5 tyrosine phosphorylation but this effect was delayed relative to the inhibition of STAT3 phosphorylation with complete inhibition not occurring until 60–120 min.

Withacnistin inhibits EGF- and IL-6-stimulated EGFR and gp130 receptor recruitment and subsequent tyrosine phosphorylation of STAT3 and STAT5. (A) MDA-MB-468 cells were serum starved for 48 h and pretreated with Wit for the indicated times then stimulated with EGF at 100 ng ml−1 for 10 min. Cells were then harvested and run for western blot analysis as described under Materials and Methods. (B) MDA-MB-468 cells were serum starved for 24 h and pretreated with Wit at increasing concentrations for 5 min, and then stimulated with EGF at 100 ng ml−1 for 30 min. Cells were then harvested and run for co-immunoprecipitation analysis as described under Materials and Methods. (C) U266 and (D) RPMI 8226 cells were serum starved for 24 h and pretreated with Wit for 5 min, and then stimulated with IL-6 at 10 ng ml−1 for 30 min. S3I designates S3I-1757, a control STAT3–EGFR association inhibitor. ‘C’ designates non-starved cells growing in 10% serum. The data in A–D are representative of three independent experiments each.
The fact that Wit required only a few minutes of treatment to have its effects and after only 10 min of EGF stimulation suggested that Wit may inhibit STAT3 and STAT5 tyrosine phosphorylation by interfering with an early step in EGF signalling. We reasoned that one possibility could be that Wit inhibits growth factor-stimulated binding of STAT3 and STAT5 to growth factor receptors. To investigate this mechanism, we performed co-immunoprecipitation studies. To this end, MDA-MB-468 cells were starved for 24 h, pretreated for 5 min with Wit, and then stimulated with EGF before immunoprecipitating with EGFR and blotting with STAT3 and STAT5 as described under Materials and Methods. Figure 1B shows that treatment of MDA-MB-468 cells with EGF in the absence of Wit stimulated STAT3 and STAT5 binding to EGFR. Pretreatment with Wit inhibited the binding of STAT3 and STAT5 to EGFR in a concentration-dependent manner. Treatment with the selective STAT3 inhibitor, S3I-1757 (Zhang et al, 2013), inhibited EGF-stimulated STAT3 but not STAT5 binding to EGFR. To determine whether Wit is able to interfere with cytokine-stimulated binding of STAT3 to their receptors, we pretreated starved U266 and RPMI8226 multiple myeloma cells with Wit, and then stimulated them with IL-6 before immunoprecipitating with the IL-6 receptor gp130 and blotting with STAT3. Figure 1C and D show that stimulation with IL-6 increased the levels of STAT3 that co-immunoprecipitated with gp130 in U266 and RPMI8226 cells, respectively, and that treatment of these cells with Wit inhibited the binding of STAT3 to gp130. Therefore, these results demonstrate that receptor–STAT interaction in intact cells was disrupted by Wit, and suggest that the ability of Wit to inhibit STAT3 and STAT5 tyrosine phosphorylation is at least in part due to inhibition of the recruitment and association of these STAT proteins with growth factor and cytokine receptors.
Wit inhibits EGF-, PDGF-, IFN-β-, IL-6-, and GM-CSF-stimulation of STAT3 and STAT5 tyrosine phosphorylation
We next sought to determine whether the effects of Wit were limited to EGF and IL-6 stimulation of STAT3 and/or STAT5 tyrosine phosphorylation. To this end, we first chose human and murine cell lines in which growth factors or cytokines are known to stimulate STAT3 phosphorylation. Starting with MDA-MB-468 cells, a human breast cancer cell line where EGF is known to induce STAT3 phosphorylation, we demonstrated as shown in Figure 2A, that Wit inhibited EGF-stimulated tyrosine phosphorylation of STAT3, with an IC50 of about 1–3 μ M. Wit, however, did not affect the ability of EGF to stimulate EGFR autophosphorylation. Furthermore, Figure 2A also shows that pretreatment with Wit of starved NIH-3T3 cells before stimulation with PDGF-BB inhibited PDGF stimulation of STAT3 tyrosine phosphorylation. Importantly, Wit did not inhibit PDGF-stimulated PDGFR tyrosine autophosphorylation. We next used the U266 human multiple myeloma cell line, where IL-6 and IFN-β stimulate STAT3 phosphorylation. Figure 2A shows that IL-6- and IFN-β-stimulated STAT3 tyrosine phosphorylation was also inhibited by Wit in U266 cells. The effect of Wit was more potent after IL-6 stimulation than after IFN-β stimulation. Finally, Figure 2A shows that Wit also inhibited potently the ability of GM-CSF to stimulate STAT3 tyrosine phosphorylation in TF-1 cells.

Withacnistin inhibits EGF-, PDGF-, IFN- β -, and GM-CSF-stimulated tyrosine phosphorylation of STAT3 and STAT5. (A) Cells were serum starved for 48 h and pretreated with Wit at increasing concentrations for 2 h, and then stimulated for 10 min with EGF at 100 ng ml−1 for 10 min (MDA-MB-468 cells), IL-6 at 10 ng ml−1 (U266 cells), GM-CSF at 2 ng ml−1 (TF-1 cells), IFN-β at 1000 U ml−1 (U266 cells), and PDGF-BB at 10 ng ml−1 (NIH-3T3 cells). The cells were then harvested and processed for western blotting with the indicated antibodies as described under Materials and Methods. (B) Cells were serum starved and pretreated with Wit as above, and then stimulated with EGF at 100 ng ml−1 for 10 min (MDA-MB-468 cells), IL-6 at 10 ng ml−1 (U266 cells), GM-CSF at 2 ng ml−1 (TF-1 cells), and PDGF-BB at 10 ng ml−1 (NIH-3T3 cells). The cells were then harvested and processed for western blotting with the indicated antibodies as described under Materials and Methods. The data in A and B are representative of at least three independent experiments for each panel.
We then expanded these studies to the effects of Wit on EGF-, PDGF-, IL-6, and GM-CSF-stimulation of STAT5 tyrosine phosphorylation as described above for STAT3. EGF, PDGF, IL-6, and GM-CSF are known to stimulate STAT5 phosphorylation in MDA-MB-468, NIH-3T3, U266, and TF-1 cells, respectively. Figure 2B shows that pretreatment of MDA-MB-468, NIH-3T3, U266 and TF-1 cells with Wit before treatment with EGF, PDGF, IL-6, and GM-CSF, respectively, inhibited growth factor and cytokine stimulation of STAT5 tyrosine phosphorylation.
Wit inhibits P-STAT3 nuclear translocation, STAT3 DNA-binding activity, transcriptional activity, and expression of STAT3 downstream genes
The translocation of phosphorylated STAT3 from cytosol to the nucleus is required for STAT3 to regulate its target genes. The fact that Wit was able to inhibit receptor–STAT3 interaction (Figure 1) suggests that it would also inhibit nuclear accumulation of tyrosine-phosphorylated STAT3. To investigate this possibility, starved MDA-MB-468 cells were pretreated for 5 min with Wit before stimulation with EGF for 30 min. The cells were then fixed and subjected to immunofluorescence staining using a specific p-Tyr-705-STAT3 primary antibody and AlexaFluor 594 secondary antibody in mounting medium containing DAPI to stain the nuclei as described under Materials and Methods. Figure 3A shows that in starved cells some tyrosine-phosphorylated STAT3 is present. In the absence of Wit, EGF stimulation resulted in a robust increase in tyrosine-phosphorylated STAT3, which was localised predominantly in the nucleus. In contrast, the levels of P-STAT3 in the nucleus were decreased in Wit-treated cells in a concentration-dependent manner, indicating that Wit inhibited EGF-stimulated P-STAT3 nuclear translocation. The control STAT3 inhibitor S3I-1757 (Zhang et al, 2013) potently inhibited EGF-stimulated P-STAT3 nuclear translocation.

Withacnistin inhibits P-STAT3 nuclear translocation and STAT3–DNA-binding activity. (A) MDA-MB-468 cells were cultured on cover slides overnight, serum starved for 24 h, pretreated with Wit at the indicated concentration for 2 h, and then stimulated with EGF at 100 ng ml−1 for 30 min. The cells were fixed using methanol for immunofluorescence analysis as described in Materials and Methods. (B) MDA-MB-468 cells were treated with either vehicle or Wit for the indicated time and the nuclear extracts were isolated from the treated cells as described in Materials and Methods. The nuclear extracts were then incubated with a biotin-labelled STAT3–DNA binding probe and the complexes were isolated using a STAT3–DNA-binding assay as described under Materials and Methods. The data in A and B are representative of two and three independent experiments.
The ability of Wit to inhibit STAT3-receptor binding, tyrosine phosphorylation, and P-STAT3 nuclear accumulation would be predicted to result in blocking STAT3–DNA binding. To evaluate this possibility, MDA-MB-468 cells were treated with vehicle or Wit at increasing concentrations for 2 h. The nuclear extracts were collected for STAT3–DNA binding activity using a STAT3 filter plate assay as described under Materials and Methods. Figure 3B shows that nuclear extracts from vehicle-treated cells contained activated STAT3 capable of binding the biotin-labelled STAT3–DNA binding probe. In contrast, the nuclear extracts from Wit-treated cells contained less-activated STAT3 capable of binding the STAT3–DNA binding probe in a concentration-dependent manner. The STAT3 inhibitor S3I-1757 (Zhang et al, 2013) was used as a control.
We next evaluated the ability of Wit to inhibit STAT3-dependent transcriptional activation using luciferase reporter assays. To this end, MDA-MB-468 cells were transiently co-transfected with a STAT3-responsive promoter-firefly luciferase reporter (pLucTKS3) and β-gal reporter used to normalise the transfection efficiency. To determine the selectivity of Wit to suppress STAT3-dependent over STAT3-independent transcriptional activation, MDA-MB-468 cells were also co-transfected with SRE promoter-renilla luciferase reporter (pLucSRE) and β-gal reporter. Figure 4A shows that compared with mock transfected cells, cells transfected with the STAT3-responsive reporter (pLucTKS3) had increased luciferase activity in the absence of drug treatment. Less luciferase activity was observed when the cells were treated with Wit. Wit inhibited STAT3-dependent but not STAT3-independent transcriptional activity as demonstrated by the minimal effect it had on SRE-driven luciferase activity (Figure 4A).

Withacnistin inhibits STAT3 transcriptional activity and expression of downstream target genes. (A) MDA-MB-468 cells were transiently transfected with pLucSRE or pLucTKS3 along with β-gal reporter genes and were then treated with vehicle or Wit as indicated. The cytosolic extracts were prepared and analysed for luciferase activity as described in Materials and Methods. C and M designate control and mock transfection, respectively. (B) MDA-MB-468 cells were treated for 24 h with the indicated concentrations of Wit and processed for western blotting with the indicated antibodies as described in Materials and Methods. S3I designates S3I-1757, a control STAT3 inhibitor. The data in A and B are representative of three and two independent experiments, respectively.
Having demonstrated that Wit inhibits STAT3 activation by blocking its binding to receptors, subsequent depletion of nuclear accumulation of P-STAT3, and loss of STAT3–DNA binding and transcriptional activity, we next determined whether the ability of STAT3 to regulate the expression of its target genes is affected by Wit. To this end, MDA-MB-468 cells were treated with vehicle and increasing concentrations of Wit and then processed for western blotting as described under Materials and Methods. Figure 4B shows that as Wit inhibited the phosphorylation of STAT3 and STAT5 in a concentration-dependent manner starting at 3 μ M; the expression of STAT3 target genes, such as the anti-apoptotic proteins Bcl-xL and Mcl-1, was also inhibited.
Treatment with Wit induces cancer cell apoptosis, inhibits cancer cell invasion and anchorage-dependent and -independent growth
Having determined that Wit prevents the activation of STAT3 by blocking its ability to associate with the EGF receptor in MDA-MB-468 cells, we next sought to determine whether the inhibition of STAT3 signalling with Wit leads to inhibition of malignant transformation. To this end, we determined the effects of Wit on apoptosis, anchorage-dependent and -independent growth, invasion, and tumour growth in vivo. Figure 5A shows that in MDA-MB-468 cells, Wit induced apoptosis (TUNEL-positive cells) in a concentration-dependent manner. We next determined the effect of Wit on anchorage-dependent and -independent growth by MTT and soft agar assays, respectively. Cell invasion assays were performed in the BD BioCoat Matrigel Invasion Chamber as described under Materials and Methods. Figure 5B shows that Wit inhibited the ability of MDA-MB-468 cells to invade in a dose-dependent manner with an IC50 value of 3 μ M. Similarly, Wit also inhibited the ability of MDA-MB-468 cells to grow in soft agar, but the effects of Wit on soft agar growth were more potent than those on invasion (Figure 4C). This potent effect of Wit was also seen in another cell line, the lung cancer cell line A549 (Figure 5D). Wit also inhibited the ability of MDA-MB-468 cells to grow in an anchorage-dependent manner in plastic (Supplementary Figure S1).

Withacnistin induces cancer cell apoptosis, inhibits cell invasion and anchorage-independent growth, and causes breast tumour regression in ErbB2 transgenic mice. (A) MDA-MB-468 cells were treated with Wit at the indicated concentration as described in Materials and Methods. The cells were fixed and the TUNEL assay was performed to determine apoptosis as described under Materials and Methods. The fold apoptosis induction was calculated by dividing the % of TUNEL-positive cells in the Wit-treated samples by those in the vehicle-treated samples. The % of TUNEL-positive cells in the vehicle-treated samples was 2.6±1.1%. (B) MDA-MB-468 cells were seeded over Matrigel in the top chamber of invasion well inserts. Cells were treated for 48 h with vehicle or Wit, and the invaded cells were fixed with methanol, stained with crystal violet, and photographed as described under Materials and Methods. MDA-MB-468 (C) and A549 (D) cells were seeded into 12-well plates in regular growth media containing 0.3% agar; Wit was added the following day, and colonies were allowed to grow for 3–4 weeks as described in Materials and Methods. The data in A–D are representative of three independent experiments each. (E) The sizes of the breast tumours from the ErbB2 transgenic mice were followed by orthogonal measurements 2–3 times per week, and the tumour volume calculated using the formula: V=(a2b)/2, where ‘a’ and ‘b’ are defined as the smaller and larger measurements, respectively. Drug treatment was initiated when the tumours grew to about 1200–2400 mm3 and treatment lasted up to 14 days. Wit (20 mpk per day) was administered i.p. into tumour-bearing mice. The percent change in tumour volume was calculated based on the tumour volume on the day of measurement relative to that on the day of initiation of treatment (T0—see Supplementary Table S1) for each mouse tumour.
Wit significantly induces breast tumour regression in ErbB2 transgenic mice
We next determined the effect of Wit on breast tumour growth in vivo in a transgenic mouse model in which mammary tumour development is driven by ErbB2, a RTK known to activate both STAT3 and STAT5. To this end, we treated these transgenic mice i.p. either with vehicle or Wit (20 mpk per day) and determined the tumour volume every other day as described under Materials and Methods. The percent change in tumour volume was calculated for each mouse based on the tumour volume on the last day of treatment (Tf—see Supplementary Table S1) relative to that on the day of initiation of treatment (T0—see Supplementary Table S1). As shown in Figure 5E; Supplementary Table S1, on average, tumours from mice treated with vehicle grew by 48.6±19.6%, whereas those from mice treated with Wit regressed by an average of 22.5±7.2% over the treatment period (P=0.0032) (Figure 5E; Supplementary Table S1). The treatments with vehicle and Wit did not result in any significant changes in body weight suggesting that Wit was non-toxic (Supplementary Table S2).
Discussion
Tyrosine phosphorylation of STAT3 and STAT5, which results in their activation, requires the rapid recruitment of these STAT proteins to growth factor and cytokine receptors following stimulation of cells with growth factors such as EGF or cytokines such as IL-6 (Darnell, 1997; Yu and Jove, 2004; Yu et al, 2009). For example, EGF binding to EGFR results in receptor dimerisation and cross-phosphorylation on tyrosine residues, which serve as binding sites for the recruitment of STAT3 and STAT5 to EGFR, and subsequent phosphorylation of the STAT proteins by this RTK (Shao et al, 2003; Ferbeyre and Moriggl, 2011). Similarly, IL-6 binding to its receptor gp130 results in recruitment of STAT proteins as well as non-RTKs Src and JAK that phosphorylate STAT3 and STAT5 (Darnell, 2002; Schust and Berg, 2004; Schust et al, 2006; Ferbeyre and Moriggl, 2011). In the work described here, we have discovered that the natural product small-molecule Wit inhibits the tyrosine phosphorylation of STAT3 and STAT5 by blocking the EGF- and IL-6-stimulated binding of EGFR and gp130 to STAT3 and STAT5. The fact that only a few minutes of treatment were necessary for Wit to rapidly inhibit EGF stimulation of P-STAT3 and P-STAT5 is consistent with interference with this early step of STAT/receptor binding. Furthermore, the finding that Wit did not inhibit EGF-stimulated EGFR tyrosine cross-phosphorylation indicates that Wit does not inhibit earlier upstream steps of growth factor receptor activation such as EGF binding to its receptor or the tyrosine kinase activity of EGFR. Therefore, Wit suppresses STAT3 and STAT5 tyrosine phosphorylation by inhibiting the recruitment of STAT3 and STAT5 to EGFR, downstream of EGF binding to its receptor, and the subsequent EGFR tyrosine kinase activation. Wit’s ability to block the recruitment of STAT3 and STAT5 to EGFR and gp130 could be due to direct binding of Wit to the STAT proteins and/or the receptors, but confirmation of this would require physical methods such as X-ray crystallography, nuclear magnetic resonance, ITC microcalorimetry or SPR Biacore. Alternatively, Wit could bind and interfere with a scaffolding protein required for receptor signalling complex formation. Chemical proteomic approaches involving mobilising Wit onto an affinity solid matrix and identifying the bound proteins from cellular lysates by mass spectrometry may be required for identifying the biochemical target/s for Wit that are involved in its ability to interfere with STAT-receptor binding.
The demonstration that Wit inhibits the activation of both STAT3 and STAT5 following stimulation with a multitude of growth factors and cytokines (e.g., EGF, PDGF, IL-6, GM-CSF, and INF-β) suggests that this natural product may be effective against a broad spectrum of human tumours. Indeed, a large number of human cancers harbour persistently tyrosine-phosphorylated, hyperactivated STAT3 and/or STAT5 due to overexpression or mutation of RTKs and non-RTKs such as EFGR, PDGFR, BCR-Abl, Src, and JAK2. For example, EGFR is commonly overexpressed and contributes to the malignancy of a variety of human cancers, notably lung and breast cancers (Sharma et al, 2007; Arora et al, 2008), while PDGF/PDGFR and IL-6/gp130 autocrine loops have pivotal roles in glioblastoma (Dunn et al, 2012) and multiple myeloma (Coluccia et al, 2008) malignancies, respectively. Furthermore, hyperactivation of the non-RTKs Abl (through the BCR–Abl fusion) and JAK2 (through mutation) contributes to several haematological malignancies (Harry et al, 2012). Finally, the GM-CSF activation of the JAK/STAT pathways has been implicated in angiogenesis, a step required for the survival of the majority of human tumours (Faderl et al, 2003).
The ability of Wit to interfere with STAT/receptor binding and subsequent tyrosine phosphorylation predicts that it would inhibit their biological activities. Indeed, Wit was very effective at inhibiting STAT3 nuclear translocation, DNA binding, transcriptional activity, and regulation of target genes. Furthermore, inhibition of STAT3 activation by Wit resulted in induction of apoptosis and inhibition of anchorage-independent growth and invasion, malignant hallmarks to which STAT3 has a known contribution. Finally, Wit treatment also induced regression of ErbB2-driven mammary tumours in an in vivo transgenic mouse model. This is an extremely important finding as ErbB2-driven cancers are known to require STAT3 and STAT5 (Hawthorne et al, 2009; Ward et al, 2013). Furthermore, ErbB2 overexpression is prevalent in tumours from breast cancer patients, and is associated with tumour aggressiveness, resistance to therapy, and poor patient survival (Hynes and Lane, 2005).
In summary, we have discovered that the small-molecule natural product Wit inhibits the recruitment of STAT3 and STAT5 to growth factor and cytokine receptors, tyrosine phosphorylation, nuclear translocation, and DNA binding, resulting in STAT3 and STAT5 inactivation and inhibition of malignant transformation both in cultured cells as well as in vivo. These findings warrant further advanced preclinical studies of Wit as an anticancer drug with the potential of broadening the treatment spectrum of human cancers that harbour persistently activated STAT3 and/or STAT5.
Change history
26 August 2014
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Arora P, Cuevas BD, Russo A, Johnson GL, Trejo J (2008) Persistent transactivation of EGFR and ErbB2/HER2 by protease-activated receptor-1 promotes breast carcinoma cell invasion. Oncogene 27 (32): 4434–4445.
Balasis ME, Forinash KD, Chen YA, Fulp WJ, Coppola D, Hamilton AD, Cheng JQ, Sebti SM (2011) Combination of farnesyltransferase and Akt inhibitors is synergistic in breast cancer cells and causes significant breast tumor regression in ErbB2 transgenic mice. Clin Cancer Res 17 (9): 2852–2862.
Bar-Natan M, Nelson EA, Walker SR, Kuang Y, Distel RJ, Frank DA (2012) Dual inhibition of Jak2 and STAT5 enhances killing of myeloproliferative neoplasia cells. Leukemia 26 (6): 1407–1410.
Chen J, Nikolovska-Coleska Z, Yang CY, Gomez C, Gao W, Krajewski K, Jiang S, Roller P, Wang S (2007) Design and synthesis of a new, conformationally constrained, macrocyclic small-molecule inhibitor of STAT3 via 'click chemistry'. BioorgMed Chem Lett 17 (14): 3939–3942.
Coluccia AM, Cirulli T, Neri P, Mangieri D, Colanardi MC, Gnoni A, Di Renzo N, Dammacco F, Tassone P, Ribatti D, Gambacorti-Passerini C, Vacca A (2008) Validation of PDGFRbeta and c-Src tyrosine kinases as tumor/vessel targets in patients with multiple myeloma: preclinical efficacy of the novel, orally available inhibitor dasatinib. Blood 112 (4): 1346–1356.
Coppo P, Dusanter-Fourt I, Millot G, Nogueira MM, Dugray A, Bonnet ML, Mitjavila-Garcia MT, Le Pesteur D, Guilhot F, Vainchenker W, Sainteny F, Turhan AG (2003) Constitutive and specific activation of STAT3 by BCR-ABL in embryonic stem cells. Oncogene 22 (26): 4102–4110.
Dai L, Cheng L, Zhang X, Jiang Q, Zhang S, Wang S, Li Y, Chen X, Du T, Yang Y, Tian H, Fan P, Yan N, Wei Y, Deng H (2011) Plasmid-based STAT3-siRNA efficiently inhibits breast tumor growth and metastasis in mice. Neoplasma 58 (6): 538–547.
Darnell JE Jr (1997) STATs and gene regulation. Science 277 (5332): 1630–1635.
Darnell JE Jr (2002) Transcription factors as targets for cancer therapy. Nat Rev Cancer 2 (10): 740–749.
Debnath B, Xu S, Neamati N (2012) Small molecule inhibitors of signal transducer and activator of transcription 3 (Stat3) protein. J Med Chem 55 (15): 6645–6668.
Deepak Mundkinajeddu, Laxman P Sawant, Rojison Koshy, Praneetha Akunuri, Vineet Kumar Singh, Anand Mayachari, Maged HM Sharaf, Murali Balasubramanian, Amit Agarwal (2014) Development and validation of high performance liquid chromatography method for simultaneous estimation of flavonoid glycosides in Withania somnifera aerial parts. ISRN Anal Chem doi:10.1155/2014/351547.
Dunn GP, Rinne ML, Wykosky J, Genovese G, Quayle SN, Dunn IF, Agarwalla PK, Chheda MG, Campos B, Wang A, Brennan C, Ligon KL, Furnari F, Cavenee WK, Depinho RA, Chin L, Hahn WC (2012) Emerging insights into the molecular and cellular basis of glioblastoma. Genes Dev 26 (8): 756–784.
Faderl S, Harris D, Van Q, Kantarjian HM, Talpaz M, Estrov Z (2003) Granulocyte-macrophage colony-stimulating factor (GM-CSF) induces antiapoptotic and proapoptotic signals in acute myeloid leukemia. Blood 102 (2): 630–637.
Ferbeyre G, Moriggl R (2011) The role of Stat5 transcription factors as tumor suppressors or oncogenes. Biochim Biophys Acta 1815 (1): 104–114.
de Groijourdy R (1864) El MBdico BotAnico Criollo. F Brachet: Paris.
Harry BL, Eckhardt SG, Jimeno A (2012) JAK2 inhibition for the treatment of hematologic and solid malignancies. Expert Opin Investig Drugs 21 (5): 637–655.
Haussler SF (1914) Schweiz Apoth-Ztg 62 (260): 275.
Hawthorne VS, Huang WC, Neal CL, Tseng LM, Hung MC, Yu D (2009) ErbB2-mediated Src and signal transducer and activator of transcription 3 activation leads to transcriptional up-regulation of p21Cip1 and chemoresistance in breast cancer cells. Mol Cancer Res 7 (4): 592–600.
Hynes NE, Lane HA (2005) ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer 5 (5): 341–354.
Kupchan SM AW, Bollinger P, Doskotch RW, Smith RM, Renauld JA, Schnoes HK, Burlingame AL, Smith DH (1969) Tumor inhibitors. XXXIX. Active principles of Acnistus arborescens. Isolation and structural and spectral studies of withaferin A and withacnistin. J Org Chem 34 (12): 3858–3866.
Malin S, McManus S, Cobaleda C, Novatchkova M, Delogu A, Bouillet P, Strasser A, Busslinger M (2010) Role of STAT5 in controlling cell survival and immunoglobulin gene recombination during pro-B cell development. Nat Immunol 11 (2): 171–179.
Mandal PK, Gao F, Lu Z, Ren Z, Ramesh R, Birtwistle JS, Kaluarachchi KK, Chen X, Bast RC Jr, Liao WS, McMurray JS (2011) Potent and selective phosphopeptide mimetic prodrugs targeted to the Src homology 2 (SH2) domain of signal transducer and activator of transcription 3. J Med Chem 54 (10): 3549–3563.
Maritano D, Sugrue ML, Tininini S, Dewilde S, Strobl B, Fu X, Murray-Tait V, Chiarle R, Poli V (2004) The STAT3 isoforms alpha and beta have unique and specific functions. Nat Immunol 5 (4): 401–409.
Nelson EA, Walker SR, Weisberg E, Bar-Natan M, Barrett R, Gashin LB, Terrell S, Klitgaard JL, Santo L, Addorio MR, Ebert BL, Griffin JD, Frank DA (2011) The STAT5 inhibitor pimozide decreases survival of chronic myelogenous leukemia cells resistant to kinase inhibitors. Blood 117 (12): 3421–3429.
Page BD, Ball DP, Gunning PT (2011) Signal transducer and activator of transcription 3 inhibitors: a patent review. Expert Opin Ther Pat 21 (1): 65–83.
Page BD, Khoury H, Laister RC, Fletcher S, Vellozo M, Manzoli A, Yue P, Turkson J, Minden MD, Gunning PT (2012) Small molecule STAT5-SH2 domain inhibitors exhibit potent antileukemia activity. J Med Chem 55 (3): 1047–1055.
Schust J, Berg T (2004) A high-throughput fluorescence polarization assay for signal transducer and activator of transcription 3. Anal Biochem 330 (1): 114–118.
Schust J, Sperl B, Hollis A, Mayer TU, Berg T (2006) Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem Biol 13 (11): 1235–1242.
Shao H, Cheng HY, Cook RG, Tweardy DJ (2003) Identification and characterization of signal transducer and activator of transcription 3 recruitment sites within the epidermal growth factor receptor. Cancer Res 63 (14): 3923–3930.
Sharma SV, Bell DW, Settleman J, Haber DA (2007) Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer 7 (3): 169–181.
Sinibaldi D, Wharton W, Turkson J, Bowman T, Pledger WJ, Jove R (2000) Induction of p21WAF1/CIP1 and cyclin D1 expression by the Src oncoprotein in mouse fibroblasts: role of activated STAT3 signaling. Oncogene 19 (48): 5419–5427.
Sun J, Blaskovich MA, Jove R, Livingston SK, Coppola D, Sebti SM (2005) Cucurbitacin Q: a selective STAT3 activation inhibitor with potent antitumor activity. Oncogene 24 (20): 3236–3245.
Sun J, Blaskovich MA, Jove R, Livingston SK, Coppola D, Sebti SM (2008) Cucurbitacin Q: a selective STAT3 activation inhibitor with potent antitumor activity. Oncogene 27: 1344.
Takeda K, Noguchi K, Shi W, Tanaka T, Matsumoto M, Yoshida N, Kishimoto T, Akira S (1997) Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc Natl Acad Sci USA 94 (8): 3801–3804.
Wang T, Niu G, Kortylewski M, Burdelya L, Shain K, Zhang S, Bhattacharya R, Gabrilovich D, Heller R, Coppola D, Dalton W, Jove R, Pardoll D, Yu H (2004) Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat Med 10 (1): 48–54.
Ward TM, Iorns E, Liu X, Hoe N, Kim P, Singh S, Dean S, Jegg AM, Gallas M, Rodriguez C, Lippman M, Landgraf R, Pegram MD (2013) Truncated p110 ERBB2 induces mammary epithelial cell migration, invasion and orthotopic xenograft formation, and is associated with loss of phosphorylated STAT5. Oncogene 32 (19): 2463–2474.
Yu H, Jove R (2004) The STATs of cancer—new molecular targets come of age. Nat Rev Cancer 4 (2): 97–105.
Yu H, Pardoll D, Jove R (2009) STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer 9 (11): 798–809.
Zhang X, Sun Y, Pireddu R, Yang H, Urlam MK, Lawrence HR, Guida WC, Lawrence NJ, Sebti SM (2013) A novel inhibitor of STAT3 homodimerization selectively suppresses STAT3 activity and malignant transformation. Cancer Res 73 (6): 1922–1933.
Zhang X, Yue P, Fletcher S, Zhao W, Gunning PT, Turkson J (2010) A novel small-molecule disrupts Stat3 SH2 domain-phosphotyrosine interactions and Stat3-dependent tumor processes. Biochem Pharmacol 79 (10): 1398–1409.
Zhang X, Yue P, Page BD, Li T, Zhao W, Namanja AT, Paladino D, Zhao J, Chen Y, Gunning PT, Turkson J (2012) Orally bioavailable small-molecule inhibitor of transcription factor Stat3 regresses human breast and lung cancer xenografts. Proc Natl Acad Sci USA 109 (24): 9623–9628.
Acknowledgements
This work was funded in part by NIH grant RO1-CA098473. We would like to thank the Microscopy Core at the Moffitt Cancer Center for their cooperation and expertise. We also thank Maria Balasis for careful reading of the manuscript as well as Dr David Newman from the National Cancer Institute Developmental Therapeutics Program for providing the Withacnistin.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Zhang, X., Blaskovich, M., Forinash, K. et al. Withacnistin inhibits recruitment of STAT3 and STAT5 to growth factor and cytokine receptors and induces regression of breast tumours. Br J Cancer 111, 894–902 (2014). https://doi.org/10.1038/bjc.2014.349
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2014.349

{kind=link}
{kind=link}
{kind=link}