
Abstract
Background:
Patients with malignant pleural effusions (MPEs) generally have advanced disease with poor survival and few therapeutic options. Cells within MPEs may be used to stratify patients for targeted therapy. Targeted therapy with poly(ADP ribose) polymerase inhibitors (PARPi) depends on identifying homologous recombination DNA repair (HRR)-defective cancer cells. We aimed to determine the feasibility of assaying HRR status in MPE cells.
Methods:
A total of 15 MPE samples were collected from consenting patients with non-small-cell lung cancer (NSCLC), mesothelioma and ovarian and breast cancer. Primary cultures were confirmed as epithelial by pancytokeratin, and HRR status was determined by the detection of γH2AX and RAD51 foci following a 24-h exposure to rucaparib, by immunofluorescence microscopy. Massively parallel next-generation sequencing of DNA repair genes was performed on cultured MPE cells.
Results:
From 15 MPE samples, 13 cultures were successfully established, with HRR function successfully determined in 12 cultures. Four samples – three NSCLC and one mesothelioma – were HRR defective and eight samples – one NSCLC, one mesothelioma, one sarcomatoid, one breast and four ovarian cancers – were HRR functional. No mutations in DNA repair genes were associated with HRR status, but there was probable loss of heterozygosity of FANCG, RPA1 and PARP1.
Conclusions:
HRR function can be successfully detected in MPE cells demonstrating the potential to stratify patients for targeted therapy with PARPi.
Similar content being viewed by others


Main
Malignant pleural effusions (MPEs), characterised by the accumulation of pleural fluid containing malignant cells, usually indicate advanced or disseminated disease with a poor median survival of between 3 and 12 months, depending on the type and stage of cancer (Roberts et al, 2010). The most common cause of MPE is lung cancer, which accounts for ∼30–40% of cases, followed by breast and ovarian cancers and lymphoma, with mesothelioma being the predominant cause of primary pleural neoplasm associated with pleural effusions (Roberts et al, 2010; Kastelik, 2013; Zarogoulidis et al, 2013). Modern cancer therapy depends on patient stratification using predictive biomarkers. The cancer cells in MPEs may be used to stratify patients for appropriate therapy. For example, those from patients with non-small-cell lung cancer (NSCLC) can be used to assess the mutational status of epidermal growth factor receptor (EGFR) for EGFR-targeted therapy with gefitinib (Hung et al, 2006).
Targeted therapy with poly(ADP ribose) polymerase inhibitors (PARPi), which are novel agents selective for cancers with dysfunctional homologous recombination DNA repair (HRR) (Bryant et al, 2005; Farmer et al, 2005), depends on the identification of HRR dysfunction. PARPi have promising anticancer activity with minimal toxicity in clinical trials in patients carrying mutations in BRCA1/2, which encode key proteins in the HRR pathway (Fong et al, 2009; O'Shaughnessy et al, 2011; De Bono et al, 2013; Kaufman et al, 2013; Ledermann et al, 2013; Michie et al, 2013). However, HRR is a multifactorial process, and screening for BRCA mutations alone is likely to underestimate the proportion of cancers with HRR defects that could potentially benefit from PARPi therapy (McCabe et al, 2006).
There is a need to develop indicators of HRR defects that may be used as predictive biomarkers for PARPi sensitivity. Different approaches to develop these are currently underway, such as integrated genomic analysis of HRR genes and aCGH to identify genomic instability (CGRN, 2011; Vollebergh et al, 2011, 2012). Alternatively, HRR function may be assessed by the ability to form RAD51 foci. RAD51 is a key protein in HRR, and its relocation onto damaged DNA is necessary for the obligatory strand-invasion step of HRR. The RAD51 focus assay correctly identified cells defective in HRR owing to BRCA1/2 mutation and BRCA1 epigenetic silencing (Drew et al, 2011b). Further application of this assay to primary cultures of epithelial ovarian cancer derived from ascitic fluid demonstrated that ∼50% had dysfunctional HRR. Importantly, RAD51 focus formation correlated with ex vivo PARPi sensitivity in >90% of cases and was an independent prognostic indicator of survival following platinum-based therapy (Mukhopadhyay et al, 2010, 2012). The RAD51 focus assay has also identified HRR defects in other patient-derived tissues: cultured breast and NSCLC biopsies, and acute myeloid leukaemia cells (Willers et al, 2009; Birkelbach et al, 2013; Gaymes et al, 2013). It has even been extended to FFPE blocks of breast tumour biopsies from patients undergoing neoadjuvant chemotherapy (Graeser et al, 2010).
The aim of this feasibility study was to extend the application of the RAD51-based HRR functional assay to primary cultures derived from MPEs to estimate the frequency of HRR defects in this population and to potentially identify those patients for whom PARPi therapy could be an option. We show that it is a viable assay for this tissue type and identified HRR defects in NSCLC and mesothelioma samples from patients. In addition, we have undertaken genetic analysis of a selection of HRR-competent and HRR-defective MPE samples; however, this did not reveal any trend in pathogenic/rare genetic variants or probable loss of heterozygosity (pLoH).
Materials and Methods
Chemicals and reagents
All routine and tissue culture chemicals and reagents were obtained from Sigma Aldrich (Poole, UK) unless otherwise stated. Rucaparib was a kind gift from Zdenek Hostomsky, Pfizer GRD (La Jolla, CA, USA).
Tissue collection and development of primary cultures
Ethical approval and specific consent were obtained for the collection of clinical material and patient data (REC 12/NW/0202). Primary cultures were derived from MPEs from cancer patients undergoing routine thoracocentesis at hospitals in Newcastle upon Tyne and Gateshead. MPE samples were transported to the laboratory and processed immediately and in accordance with the regulations of the Human Tissue Act 2004 (UK) and local guidelines.
Briefly, 50 ml of MPE was centrifuged and the cells and debris were concentrated in 5 ml of the supernatant, mixed with 5 ml of RPMI-1640, supplemented with 20% fetal bovine serum (FBS) and 100 U ml−1 penicillin and 100 μg ml−1 streptomyocin, and cultured in a 25-cm3 tissue culture flask at 37 °C with 5% CO2 for 3–12 days. Culture medium was replenished every 7 days, and cultures were passaged at a confluency of 60–80%; further culturing was carried out with RPMI-1640 (10% FBS, 100 U ml−1 penicillin/streptomyocin) alone. All experiments were carried out on early-passage cultures (<4).
Immunofluorescence assays
Cells were seeded onto coverslips at a density of 0.25–1 × 105 cells ml−1 depending on cell numbers and incubated for 24–48 h in a 37 °C incubator with 5% CO2. To confirm epithelial cell origin, cells were fixed and permeabilised with ice-cold methanol, washed with PBS with 0.4% Triton-X-100, blocked with PBS containing 2% (w/v) bovine serum albumin (BSA) and then incubated with 1 : 100 FITC-conjugated anti-pancytokeratin antibody (Merck Millipore, Watford, UK). Coverslips were mounted onto slides with mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI) (Vectashield, Peterborough, UK).
For the HRR functional assay, DNA replication fork collapse was induced with 10 μ M of the PARPi rucaparib (AG-014699) for 24 h before fixation and permeabilisation, as described previously (Mukhopadhyay et al, 2010). Coverslips were blocked with PBS containing 2% BSA (w/v), 10% skimmed milk powder (w/v) and 10% goat serum (v/v), and then incubated overnight at 4 °C with 1 : 100 anti-RAD51 antibody (PC130 Calbiochem, Merk Millipore). Coverslips were washed in PBS and then incubated with anti-phospho-Histone H2A.X (Ser139) (clone JBW301, Merck Millipore) diluted to 1 : 1000 in PBS containing 2% BSA (w/v). After washing with PBS containing 0.4% Triton-X-100, coverslips were incubated with secondary antibodies Alexa Fluor 546 Goat anti-mouse and Alexa Fluor 488 Goat anti-rabbit antibodies (Invitrogen, Life Technologies, Paisley, UK), diluted to 1 : 1000 and incubated in the dark to visualise γH2AX and RAD51, respectively. Coverslips were mounted as described earlier.
Fluorescence microscopy was conducted using a Leica DMR microscope (Leica microsystems GmbH, Wetzlar, Germany) or a confocal microscope. The number of γH2AX and RAD51 foci was analysed in >50 cells per condition by using the ImageJ software with the PZFociEZ macro (www.pzfociez.com).
Next-generation sequencing
Genomic DNA extracted from cancer cells cultured from MPEs was sheared to a mean length of 500 bp using nitrogen nebulisation. A custom-made Nimblegen SeqCap EZ library (Nimblegen, Madison, WI, USA) was used to enrich for the sequence of interest. This comprised the exonic regions of >180 DNA repair genes and limited intronic material. Captured sequence was subjected to massively parallel next-generation DNA sequencing by using the Roche 454 GS FLX platform (454 Life Science, Branford, CT, USA). A bespoke analysis pipeline was used to identify pathogenic changes and potential variants of interest (single nucleotide polymorphisms with <1% population prevalence) across our target genes. pLoH – that is, loss of genetic variation, rather than copy number – was evaluated by plotting the percentage reads for every detected variant across individual genes. Tracts of contiguous variants detected at >80% reads were considered pLoH.
Statistical analyses
GraphPad Prism (version 6.00, San Diego, CA, USA) was used for analyses. Univariate analysis of overall survival (OS) was carried out by generating a Kaplan–Meier survival curve; statistical significance was derived from Mantel–Cox log-rank tests for statistical significance.
Results
Establishing primary cultures from pleural effusion samples
MPE samples were collected from patients diagnosed with different cancers (ovarian, breast, NSCLC and mesothelioma); patient characteristics are described in Table 1. A total of 15 MPE samples were collected and primary cultures were established (denoted primary pleural effusion (PPE)) by using an optimised methodology based on that established previously for ascites cells (Mukhopadhyay et al, 2010). PPE cultures were established for 13 out of 15 samples; 2 samples (PPE004 and PPE005, ovarian cancers) were contaminated from the outset of culturing and were not assessed further. PPE cultures grew as an adherent monolayer in discreet patches; the majority of cells had a polygonal shape and displayed a cobblestone-like appearance at confluency (Figure 1A). Epithelial cell cultures were confirmed with pancytokeratin immunofluorescence; all PPE cultures had >90% cells with a positive pancytokeratin phenotype (Figure 1B).

Pleural effusion primary cultures (A) Primary cultures established from pleural effusion samples grew as a monolayer exhibiting a polygonal cell morphology and a cobblestone-like appearance at confluency. (B) Epithelial cell growth in primary cultures assessed by pancytokeratin staining; cell nuclei are visualised with DAPI. Cultures with >90% positive cells were analysed further. Example images are taken from PPE007, a pleural effusion from a breast cancer.
Determining HRR status in primary cultures
The HRR status of primary cultures was assessed using the immunofluorescence-based γH2AX and RAD51 assay. A greater than 2-fold increase in γH2AX foci was taken as confirmation that stalled/collapsed replication forks and/or DNA double-strand breaks (DSB) had been generated. Cells were defined as HRR competent if a ⩾2-fold increase in RAD51 foci was also observed and HRR defective if there was no significant increase in RAD51 foci (Figure 2A). Using these thresholds, DNA damage was induced in 12 out of 13 samples analysed. DNA DSBs were not induced in PPE011 (ovarian cancer). Of the remaining samples, eight were identified as HRR competent (four ovarian cancer, one mesothelioma, one sarcomatoid lung carcinoma and one breast cancer) and four were found to be HRR defective (three NSCLC and one mesothelioma) (Figure 2B, Table 2).

HRR status in primary cultures. (A) Example immunofluorescence microscopy in primary cultures following DSB induction with a 24-h exposure to rucaparib. In HRR-competent cultures (PPE012, NSCLC), increased levels of γH2AX (indicating DSB) and RAD51 (indicating HRR) foci are seen in the nucleus following rucaparib treatment; however, in cultures with dysfunctional HRR (PPE003, NSCLC), only increased levels of γH2AX foci are seen following rucaparib treatment (+) compared with untreated control (−). (B) HRR status of primary cultures. Average number of γH2AX and RAD51 foci per cell was determined by foci counting (ImageJ). Foci numbers were normalised to the control and expressed as a fold induction following rucaparib treatment. A 2-fold induction (dashed line) was set as the threshold for γH2AX and RAD51 induction. Data are representative of 2–3 independent experiments (PPE002, 003, 007, 008, 009, 012, 014), with error bars indicating the s.e.m., or a single experiment (PPE001, 006, 010, 013, 015) where sample sizes were small or cultures stopped proliferating at an early passage.
Clinical data
Defects in HRR render cells highly sensitive to platinum agents, and, in our previous studies, patients with ovarian cancer identified as HRR defective had greater than OS following platinum-based therapy (Mukhopadhyay et al, 2012). To investigate whether the HRR status of the PPEs in this cohort of patients had any bearing on response to therapy or survival, clinical data were collected. Of the 12 patients in whom tumour HRR status was determined, only the patients with ovarian cancer and breast cancer and one of the patients with mesothelioma received anticancer therapy (Table 2). None of the patients with NSCLC were fit enough to receive platinum-based therapy, but the patient with the HRR-defective mesothelioma received platinum-based therapy. There was no statistical difference in the OS between the HRR functional and dysfunctional patients (data not shown).
Genetic investigation
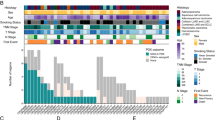
The exonic regions of >180 DNA repair genes were captured and subjected to massively parallel sequencing (full list of genes studied provided in Supplementary Figure 1). Known pathogenic variants and variants with a reported population prevalence <1% were identified. No pathogenic/rare genetic variants or pLoH were observed recurrently in either HRR-competent or dysfunctional PPE cultures (Figure 3). Our results do not support the role of any single gene in determining HRR status in tumours generally. pLoH was restricted to HRR-defective tumours for FANCG, PARP1 and RPA1. We have previously used this simple approach to identify regions of pLoH in tumour samples that were confirmed by array comparative genomic hybridisation to be monoallelic, as a result of both interstitial duplication and deletion (data not shown). Our chosen sequencing platform does not lend itself to the evaluation of copy number in these tumours, owing to the comparatively modest read depth.

Summary of genetic data. The distribution of putative pathogenic variants (known mutations or variants with a population frequency of <1 : 1000) across the major DNA maintenance families is shown, together with probable loss of heterozygosity. As private aneuploidies may skew coverage for individual cancers, expected sequencing performance is represented by mean target coverage (exons +10 bp/−50 bp) at a depth >15 reads for diploid samples (n=13). Only two nonsense mutations were detected across the listed genes; both were heterozygous. Three genes demonstrated pLoH in HRR-defective tumours only – FANCG, RPA1 and, surprisingly, PARP1. There is no clear pattern of genetic changes associated with HRR status, although more subtle relationships may be detected using a larger sample number.
Discussion
In this feasibility study, we have shown that we are able to culture a variety of different cancer cell types from MPEs with an 80% success rate. Furthermore, we were able to determine the HRR status of 12 out of 13 samples that were successfully cultured. Therefore, cells from MPEs represent a source of patient-derived tumour material that could potentially be used to stratify patients for either PARPi or platinum-based therapy. We believe that MPEs represent a valuable resource that would otherwise be disposed of.
We have established that around 35% of PPE cultures overall were HRR dysfunctional. This relatively high frequency may reflect that the loss of a high-fidelity DNA repair pathway can generate genomic instability that is an enabling characteristic of cancer (Hanahan and Weinberg, 2011). Defective DNA repair may underlie the response of tumours to DNA-damaging anticancer therapy (Curtin, 2012). Indeed, defects in DNA repair, particularly those associated with HRR, are a common feature of adult cancers, as well as paediatric cancer syndromes (Kennedy and D'Andrea, 2006). We may even have underestimated the proportion of HRR-defective tumours, as recent evidence indicates that some NSCLC cell lines have defects in HRR downstream of RAD51 focus formation, and instead fail to resolve these foci leading to their persistence (Kommajosyula et al, 2013; Postel-Vinay et al, 2013). Adaptation of the current HRR functional assay in future studies will allow detection of defects in the resolution of RAD51.
Our previous studies, and those of others, indicate that around 50% of ovarian cancers are HRR defective (Mukhopadhyay et al, 2010; CGRN, 2011). It was therefore somewhat surprising that none of the four PPE cultures derived from ovarian cancers were HRR defective. This may be due to chance in this small sample size. Alternatively, the platinum-based therapy that the patients received (Table 2) may have led to the restoration of HRR function or selection of HRR functional clones from a heterogeneous tumour. Restoration of HRR function in cancers associated with BRCA1 and BRCA2 mutations has been observed in both the laboratory and clinical setting (Patel et al, 2012).
Interestingly, three of the four NSCLC samples analysed in this pilot study were found to have dysfunctional HRR. Evidence of HRR dysfunction in lung cancer is emerging. A recent study identified that 4 out of 16 NSCLC cell lines failed to form RAD51 foci following cisplatin-induced DNA damage and that this correlated with olaparib (a PARPi) sensitivity for 14 out of 16 cell lines (Birkelbach et al, 2013). Furthermore, this group identifies two tumours with a low RAD51 score, indicative of dysfunctional HRR, in fresh tumour tissues from 13 patients with NSCLC (Birkelbach et al, 2013). In addition, data showing mutations in HRR genes in lung cancer are accumulating. Mutations in ATM have been identified in 13 out of 188 primary lung adenocarcinomas (Ding et al, 2008), and a BRCA1 deficiency has been demonstrated in 11–19% of NSCLC patients (Paul et al, 2011). Loss of PTEN is seen in ∼4.5% of NSCLC (Jin et al, 2010), and in lung cancer cell lines PTEN loss leads to abrogation of HRR, which was associated with the suppression of lung tumour xenograft growth in mice treated with olaparib and cisplatin (Minami et al, 2013). Epigenetic alterations in key HRR proteins have also been identified, with hypermethylation of BRCA1 and FANCF being identified in lung cancers (Esteller et al, 2001; Marsit et al, 2004).
To investigate further the predictive biomarkers for HRR function in PPE cultures, massively parallel sequencing of a panel of DNA repair genes was undertaken in three competent and two dysfunctional HRR PPE cultures following sequence capture. No trends of genetic aberrations were identified. However, pLOH of PARP1, RPA1 and FANCG was observed only in dysfunctional HRR PPE cultures, the functional significance of which is unclear. However, in vertebrates, FANCG is essential for the repair of a subset of DSBs and is involved in the recruitment of FANCD2, BRCA2 and XRCC3 to these lesions (Yamamoto et al, 2003; Wilson et al, 2008; Orta et al, 2013). It is possible that pLoH involving RPA1, which is recruited to single-stranded DNA at collapsed replication forks (Hass et al, 2012), also affects HRR function. Our small sample size prevents confident detection of patterns on a biological pathway basis (for example, combinations of various HRR and BER factors generating a more complex genetic signature). In addition, there is increasing evidence for genetic and histological heterogeneity within individual tumours. It may be that contributory genetic signatures have been masked by culturing heterogeneous tumour cells from MPEs before analysis.
Lung cancer is the most common cancer, accounting for 1.6 million cancer diagnoses worldwide, and the largest cause of cancer-related deaths (CRUK, 2013). Most lung cancer diagnoses are made when the disease is at an advanced and/or metastatic stage, and prognosis is poor (<10% 5-year survival (CRUK, 2013)). Our data suggest that potentially exploitable HRR defects are relatively common in this disease. Although the growth of the PPE cells was too poor to determine ex vivo PARPi sensitivity, we would nevertheless, on the basis of accumulated evidence in a variety of tumour types, predict that patients with HRR-defective NSCLC may benefit from PARPi therapy. All of the patients with NSCLC were too ill to receive conventional chemotherapy. However, the patient with HRR-defective mesothelioma underwent several rounds of carboplatin/pemetrexed therapy (Table 2) before succumbing to the disease. On the basis of only one patient, it is impossible to establish whether this response to therapy was due to an HRR defect, although the data are encouraging.
NSCLC patients are often not eligible for platinum-based chemotherapy owing to concomitant illness. Given that PARPi are selectively toxic to HRR-defective tumour tissue while causing negligible clinical toxicity (Fong et al, 2009, 2010; Schelman et al, 2011; Drew et al, 2011a), treating NSCLC patients with a PARPi may be an attractive option. Excitingly, there are indications that PARPi may be of benefit in the treatment of NSCLC. A phase I dose escalation study of the PARP inhibitor niraparib (MK4827) included two patients with NSCLC. In one patient (carrying a BRCA2 mutation), stable disease was observed for 175 days, and in the other (with platinum-sensitive disease) for 316 days (Sandhu et al, 2013). A further application of the HRR functional assay using MPEs may be in the selection of SCLC patients for PARPi treatment; a phase I trial for the PARPi BMN 673 is currently underway and recruiting SCLC patients (NCT01286987, clinicaltrials.gov). We propose that this feasibility study may be extended to a larger cohort of cancer patients presenting with MPEs and may be used in stratifying patients, in particular NSCLC patients, to receive PARP inhibitor treatment.
Change history
01 July 2014
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Birkelbach M, Ferraiolo N, Gheorghiu L, Pfaffle HN, Daly B, Ebright MI, Spencer C, O'Hara C, Whetstine JR, Benes CH, Sequist LV, Zou L, Dahm-Daphi J, Kachnic LA, Willers H (2013) Detection of impaired homologous recombination repair in NSCLC cells and tissues. J Thorac Oncol 8 (3): 279–286.
Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T (2005) Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434 (7035): 913–917.
CGRN (2011) Integrated genomic analyses of ovarian carcinoma. Nature 474 (7353): 609–615.
CRUK (2013) Cancer Statistics Report: Cancer Mortality in the UK in 2010.
Curtin NJ (2012) DNA repair dysregulation from cancer driver to therapeutic target. Nat Rev Cancer 12 (12): 801–817.
De Bono JS, Mina LA, Gonzalez M, Curtin NJ, Want E, Henshaw JW, Chadha M, Sachdev JC, Matei D, Jameson GS, Ong M, Basu B, Wainberg ZA, Byers LA, Chugh R, Dorr A, Kaye SB, Ramanathan RK (2013) First-in-human trial of novel oral PARP inhibitor BMN 673 in patients with solid tumours. J Clin Oncol 31 (suppl): ): abstr 2580.
Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, Sougnez C, Greulich H, Muzny DM, Morgan MB, Fulton L, Fulton RS, Zhang Q, Wendl MC, Lawrence MS, Larson DE, Chen K, Dooling DJ, Sabo A, Hawes AC, Shen H, Jhangiani SN, Lewis LR, Hall O, Zhu Y, Mathew T, Ren Y, Yao J, Scherer SE, Clerc K, Metcalf GA, Ng B, Milosavljevic A, Gonzalez-Garay ML, Osborne JR, Meyer R, Shi X, Tang Y, Koboldt DC, Lin L, Abbott R, Miner TL, Pohl C, Fewell G, Haipek C, Schmidt H, Dunford-Shore BH, Kraja A, Crosby SD, Sawyer CS, Vickery T, Sander S, Robinson J, Winckler W, Baldwin J, Chirieac LR, Dutt A, Fennell T, Hanna M, Johnson BE, Onofrio RC, Thomas RK, Tonon G, Weir BA, Zhao X, Ziaugra L, Zody MC, Giordano T, Orringer MB, Roth JA, Spitz MR, Wistuba II, Ozenberger B, Good PJ, Chang AC, Beer DG, Watson MA, Ladanyi M, Broderick S, Yoshizawa A, Travis WD, Pao W, Province MA, Weinstock GM, Varmus HE, Gabriel SB, Lander ES, Gibbs RA, Meyerson M, Wilson RK (2008) Somatic mutations affect key pathways in lung adenocarcinoma. Nature 455 (7216): 1069–1075.
Drew Y, Ledermann JA, Jones A, Hall G, Jayson GC, Highley M, Rea D, Glasspool RM, Halford SER, Crosswell G, Colebrook S, Boddy AV, Curtin NJ, Plummer ER (2011a) Phase II trial of the poly(ADP-ribose) polymerase (PARP) inhibitor AG-014699 in BRCA 1 and 2-mutated, advanced ovarian and/or locally advanced or metastatic breast cancer. J Clin Oncol 29 (15_suppl): 3104.
Drew Y, Mulligan EA, Vong WT, Thomas HD, Kahn S, Kyle S, Mukhopadhyay A, Los G, Hostomsky Z, Plummer ER, Edmondson RJ, Curtin NJ (2011b) Therapeutic potential of poly(ADP-ribose) polymerase inhibitor AG014699 in human cancers with mutated or methylated BRCA1 or BRCA2. J Natl Cancer Inst 103 (4): 334–346.
Esteller M, Corn PG, Baylin SB, Herman JG (2001) A gene hypermethylation profile of human cancer. Cancer Res 61 (8): 3225–3229.
Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, Ashworth A (2005) Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434 (7035): 917–921.
Fong PC, Boss DS, Yap TA, Tutt A, Wu PJ, Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O'Connor MJ, Ashworth A, Carmichael J, Kaye SB, Schellens JHM, de Bono JS (2009) Inhibition of poly(ADP-Ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med 361 (2): 123–134.
Fong PC, Yap TA, Boss DS, Carden CP, Mergui-Roelvink M, Gourley C, De Greve J, Lubinski J, Shanley S, Messiou C, A'Hern R, Tutt A, Ashworth A, Stone J, Carmichael J, Schellens JHM, de Bono JS, Kaye SB (2010) Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J Clin Oncol 28 (15): 2512–2519.
Gaymes TJ, Mohamedali AM, Patterson M, Matto N, Smith A, Kulasekararaj A, Chelliah R, Curtin N, Farzaneh F, Shall S, Mufti GJ (2013) Microsatellite instability induced mutations in DNA repair genes CtIP and MRE11 confer hypersensitivity to poly (ADP-ribose) polymerase inhibitors in myeloid malignancies. Haematologica 98 (9): 1397–1406.
Graeser M, McCarthy A, Lord CJ, Savage K, Hills M, Salter J, Orr N, Parton M, Smith IE, Reis-Filho JS, Dowsett M, Ashworth A, Turner NC (2010) A marker of homologous recombination predicts pathologic complete response to neoadjuvant chemotherapy in primary breast cancer. Clin Cancer Res 16 (24): 6159–6168.
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144 (5): 646–674.
Hass CS, Lam K, Wold MS (2012) Repair-specific functions of replication protein A. J Biol Chem 287 (6): 3908–3918.
Hung MS, Lin CK, Leu SW, Wu MY, Tsai YH, Yang CT (2006) Epidermal growth factor receptor mutations in cells from non-small cell lung cancer malignant pleural effusions. Chang Gung Med J 29 (4): 373–379.
Jin G, Kim MJ, Jeon HS, Choi JE, Kim DS, Lee EB, Cha SI, Yoon GS, Kim CH, Jung TH, Park JY (2010) PTEN mutations and relationship to EGFR, ERBB2, KRAS, and TP53 mutations in non-small cell lung cancers. Lung Cancer 69 (3): 279–283.
Kastelik JA (2013) Management of malignant pleural effusion. Lung 191 (2): 165–175.
Kaufman B, Shapira-Frommer R, Schmulzler RK, Audeh MW, Friedlander M, Balmana J, Mitchel G, Fried G, Bowman K, Fielding A, Domchek SM, Sheba C (2013) Olaparib monotherapy in patienrs with advanced cancer and a germ-line BRCA1/2 mutation: An open-label phase II study. J Clin Oncol 31 (suppl): ): abst 11024.
Kennedy RD, D'Andrea AD (2006) DNA repair pathways in clinical practice: lessons from pediatric cancer susceptibility syndromes. J Clin Oncol 24 (23): 3799–3808.
Kommajosyula K, Cao Y, Moreau L, D'Andrea A, Shapiro GI (2013) Identification of factors governing sensitivity of NSCLC cell lines to inhibition of poly (ADP-ribose) polymerase (PARP). In Proceedings of the 104th Annual Meeting of the American Association for Cancer Research; 2013 April, pp 6–10. Washington, DC, PA, USA, Cancer Res, 73(8 Suppl): Abstract nr 1781.
Ledermann JA, Harter P, Gourley C, Friedlander M, Vergote I, Rustin GJS, Meier W, Shapira-Frommer R, Safra T, Matei D, Fielding A, Macpherson E, Dougherty B, Jurgensmeier JM, Orr M, Matulonis U (2013) Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer (SOC) and a BRCA mutation (BRCAm). J Clin Oncol 31 (suppl): ): abstr 5505.
Marsit CJ, Liu M, Nelson HH, Posner M, Suzuki M, Kelsey KT (2004) Inactivation of the Fanconi anemia/BRCA pathway in lung and oral cancers: implications for treatment and survival. Oncogene 23 (4): 1000–1004.
McCabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A, Swift S, Giavara S, O'Connor MJ, Tutt AN, Zdzienicka MZ, Smith GC, Ashworth A (2006) Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res 66 (16): 8109–8115.
Michie CO, Sandhu SK, Schelman WR, Molife LR, Wilding G, Omlin AG, Kansra V, Brooks DG, Martel RE, Kaye SB, De Bono JS, Wenham RM (2013) Final results of the pahse I trial of niraparib (MK4827), a poly(ADP)ribose polymerase (PARP) inhibitor incorporating proof of concept biomarker studies and expansion cohorts involving BRCA1/2 mutation carriers, sporadic ovarian, and castration resistant prostate cancer (CRPC). J Clin Oncol 31 (suppl): ): abstr 2513.
Minami D, Takigawa N, Takeda H, Takata M, Ochi N, Ichihara E, Hisamoto A, Hotta K, Tanimoto M, Kiura K (2013) Synergistic effect of olaparib with combination of cisplatin on PTEN-deficient lung cancer cells. Mol Cancer Res 11 (2): 140–148.
Mukhopadhyay A, Elattar A, Cerbinskaite A, Wilkinson SJ, Drew Y, Kyle S, Los G, Hostomsky Z, Edmondson RJ, Curtin NJ (2010) Development of a functional assay for homologous recombination status in primary cultures of epithelial ovarian tumor and correlation with sensitivity to poly(ADP-ribose) polymerase inhibitors. Clin Cancer Res 16 (8): 2344–2351.
Mukhopadhyay A, Plummer ER, Elattar A, Soohoo S, Uzir B, Quinn JE, McCluggage WG, Maxwell P, Aneke H, Curtin NJ, Edmondson RJ (2012) Clinicopathological features of homologous recombination-deficient epithelial ovarian cancers: sensitivity to PARP inhibitors, platinum, and survival. Cancer Res 72 (22): 5675–5682.
O'Shaughnessy J, Osborne C, Pippen JE, Yoffe M, Patt D, Rocha C, Koo IC, Sherman BM, Bradley C (2011) Iniparib plus chemotherapy in metastatic triple-negative breast cancer. N Engl J Med 364 (3): 205–214.
Orta ML, Calderon-Montano JM, Dominguez I, Pastor N, Burgos-Moron E, Lopez-Lazaro M, Cortes F, Mateos S, Helleday T (2013) 5-Aza-2′-deoxycytidine causes replication lesions that require Fanconi anemia-dependent homologous recombination for repair. Nucleic Acids Res 41 (11): 5827–5836.
Patel AG, De Lorenzo SB, Flatten KS, Poirier GG, Kaufmann SH (2012) Failure of iniparib to inhibit poly(ADP-Ribose) polymerase in vitro. Clin Cancer Res 18 (6): 1655–1662.
Paul I, Savage KI, Blayney JK, Lamers E, Gately K, Kerr K, Sheaff M, Arthur K, Richard DJ, Hamilton PW, James JA, O'Byrne KJ, Harkin DP, Quinn JE, Fennell DA (2011) PARP inhibition induces BAX/BAK-independent synthetic lethality of BRCA1-deficient non-small cell lung cancer. J Pathol 224 (4): 564–574.
Postel-Vinay S, Bajrami I, Friboulet L, Elliott R, Fontebasso Y, Dorvault N, Olaussen KA, Andre F, Soria JC, Lord CJ, Ashworth A (2013) A high-throughput screen identifies PARP1/2 inhibitors as a potential therapy for ERCC1-deficient non-small cell lung cancer. Oncogene 32 (47): 5377–5387.
Roberts ME, Neville E, Berrisford RG, Antunes G, Ali NJ (2010) Management of a malignant pleural effusion: British Thoracic Society Pleural Disease Guideline 2010. Thorax 65 (Suppl 2): ii32–ii40.
Sandhu SK, Schelman WR, Wilding G, Moreno V, Baird RD, Miranda S, Hylands L, Riisnaes R, Forster M, Omlin A, Kreischer N, Thway K, Gevensleben H, Sun L, Loughney J, Chatterjee M, Toniatti C, Carpenter CL, Iannone R, Kaye SB, de Bono JS, Wenham RM (2013) The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: a phase 1 dose-escalation trial. Lancet Oncol 14 (9): 882–892.
Schelman WR, Sandhu SK, Monreno Garcia V, Wilding G, Sun L, Toniatti C, Stroh M, Kreischer N, Carpenter CL, Molife LR, Kaye SB, De Bono JS, Wenham RM (2011) First-in-human trial of a poly(ADP)-ribose polymerase (PARP) inhibitor MK-4827 in advanced cancer patients with antitumor activity in BRCA-deficient tumors and sporadic ovarian cancers (soc). J Clin Oncol 29 (suppl): ): abstr 3102.
Vollebergh MA, Jonkers J, Linn SC (2012) Genomic instability in breast and ovarian cancers: translation into clinical predictive biomarkers. Cell Mol Life Sci 69 (2): 223–245.
Vollebergh MA, Lips EH, Nederlof PM, Wessels LF, Schmidt MK, van Beers EH, Cornelissen S, Holtkamp M, Froklage FE, de Vries EG, Schrama JG, Wesseling J, van de Vijver MJ, van Tinteren H, de Bruin M, Hauptmann M, Rodenhuis S, Linn SC (2011) An aCGH classifier derived from BRCA1-mutated breast cancer and benefit of high-dose platinum-based chemotherapy in HER2-negative breast cancer patients. Ann Oncol 22 (7): 1561–1570.
Willers H, Taghian AG, Luo CM, Treszezamsky A, Sgroi DC, Powell SN (2009) Utility of DNA repair protein foci for the detection of putative BRCA1 pathway defects in breast cancer biopsies. Mol Cancer Res 7 (8): 1304–1309.
Wilson JB, Yamamoto K, Marriott AS, Hussain S, Sung P, Hoatlin ME, Mathew CG, Takata M, Thompson LH, Kupfer GM, Jones NJ (2008) FANCG promotes formation of a newly identified protein complex containing BRCA2, FANCD2 and XRCC3. Oncogene 27 (26): 3641–3652.
Yamamoto K, Ishiai M, Matsushita N, Arakawa H, Lamerdin JE, Buerstedde JM, Tanimoto M, Harada M, Thompson LH, Takata M (2003) Fanconi anemia FANCG protein in mitigating radiation- and enzyme-induced DNA double-strand breaks by homologous recombination in vertebrate cells. Mol Cell Biol 23 (15): 5421–5430.
Zarogoulidis K, Zarogoulidis P, Darwiche K, Tsakiridis K, Machairiotis N, Kougioumtzi I, Courcoutsakis N, Terzi E, Zaric B, Huang H, Freitag L, Spyratos D (2013) Malignant pleural effusion and algorithm management. J Thorac Dis 5 (Suppl 4): S413–S419.
Acknowledgements
We gratefully acknowledge the generosity and co-operation of the patients who donated their tissue and the support of clinical and nursing staff (Dr Sarah Woolcock, Queen Elizabeth Hospital). This work was funded by Newcastle upon Tyne Hospitals NHS Foundation Trust and The Newcastle Healthcare Charity and Newcastle upon Tyne NHS Charity, to whom we are most grateful.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Patterson, M., Sutton, R., Forrest, I. et al. Assessing the function of homologous recombination DNA repair in malignant pleural effusion (MPE) samples. Br J Cancer 111, 94–100 (2014). https://doi.org/10.1038/bjc.2014.261
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2014.261
Keywords
This article is cited by
-
Poly(ADP-ribose) polymerase inhibition: past, present and future
Nature Reviews Drug Discovery (2020)
-
Use of poly ADP-ribose polymerase [PARP] inhibitors in cancer cells bearing DDR defects: the rationale for their inclusion in the clinic
Journal of Experimental & Clinical Cancer Research (2016)
