
Abstract
Background:
Cetuximab is the only targeted agent approved for the treatment of head and neck squamous cell carcinomas (HNSCC), but low response rates and disease progression are frequently reported. As the phosphoinositide 3-kinase (PI3K) and the mammalian target of rapamycin (mTOR) pathways have an important role in the pathogenesis of HNSCC, we investigated their involvement in cetuximab resistance.
Methods:
Different human squamous cancer cell lines sensitive or resistant to cetuximab were tested for the dual PI3K/mTOR inhibitor PF-05212384 (PKI-587), alone and in combination, both in vitro and in vivo.
Results:
Treatment with PKI-587 enhances sensitivity to cetuximab in vitro, even in the condition of epidermal growth factor receptor (EGFR) resistance. The combination of the two drugs inhibits cells survival, impairs the activation of signalling pathways and induces apoptosis. Interestingly, although significant inhibition of proliferation is observed in all cell lines treated with PKI-587 in combination with cetuximab, activation of apoptosis is evident in sensitive but not in resistant cell lines, in which autophagy is pre-eminent. In nude mice xenografted with resistant Kyse30 cells, the combined treatment significantly reduces tumour growth and prolongs mice survival.
Conclusions:
Phosphoinositide 3-kinase/mammalian target of rapamycin inhibition has an important role in the rescue of cetuximab resistance. Different mechanisms of cell death are induced by combined treatment depending on basal anti-EGFR responsiveness.
Similar content being viewed by others


Main
Epidermal growth factor receptor (EGFR) is a transmembrane tyrosine kinase receptor that has a central role in regulating cell division and death. An overexpression of the receptor, of its phosphorylated form or of any of its linked pathways occurs in most epithelial cancers, including head and neck squamous cell cancer (HNSCC), and it correlates with poor prognosis and resistance to anticancer therapies (Maurizi et al, 1996; Van Waes et al, 2010; Baba et al, 2012). Among different anti-EGFR agents, such as tyrosine kinase inhibitorslike gefitinib, erlotinib or lapatinib, and monoclonal antibodies (mAbs), such as cetuximab, panitumumab and zalutumumab, that were tested in the clinical setting, cetuximab is the only targeted agent approved for the treatment of HNSCC (Bianco et al, 2005; Nestor, 2010). Despite its efficacy, the clinical use of cetuximab is significantly limited by the intrinsic resistance observed in a number of patients and by the onset of acquired resistance often arising in responders. Several mechanisms of resistance to EGFR inhibitors have been elucidated to date. It has been reported that oncogenic activation of intracellular signalling downstream EGFR, including RAS/RAF/MAPK and PI3K/PTEN/AKT pathways, is an important mechanism for generating resistance to anti-EGFR mAbs. For example, activating mutations of the phosphoinositide 3-kinase catalytic subunit alpha (PIK3CA) were found likely to predict resistance to anti-EGFR mAbs in metastatic colorectal cancer (Moroni et al, 2005). However, each of these mechanisms only partially contribute to justify the lack of response to EGFR inhibitors, in preclinical models as well as in cancer patients. Therefore, the search for further determinants of resistance may enable a better selection of patients potentially responsive to anti-EGFR drugs and the development of novel therapeutic strategies to overcome EGFR resistance.
The mammalian target of rapamycin (mTOR) and the phosphoinositide 3-kinase (PI3K) signalling pathways have been found to have an important role in the pathogenesis of HNSCC (Liu et al, 2009), and data from literature suggest that PI3K antagonists are active against head and neck cancer cells (Bancroft et al, 2002). In HNSCC cell lines and tumour specimens, sensitivity to anti-EGFR therapy has been correlated with activation of phosphorylated Akt (Pernas et al, 2009). In addition, human HNSCC often display multiple alterations of the PI3K pathway, including increased PIK3CA expression, PIK3CA mutations, Akt amplification and mTOR activation (Pedrero et al, 2005; Herzog et al, 2013). The mammalian target of rapamycin is a highly conserved serine/threonine kinase downstream of the PI3K/AKT pathway; it has a crucial role in tumorigenesis (Guertin and Sabatini, 2007) and its inhibition could prevent cancer cell proliferation (Engelman et al, 2006; Shaw and Cantley, 2006; Yuan and Cantley, 2008). Despite the rapamycin analogues having been approved for the treatment of some human malignancies, such as metastatic renal cell carcinoma, breast and neuroendocrine cancers, they only account for inhibition of the target-of-rapamycin complex 1 (TORC1). Several experimental studies demonstrate that the TORC2 complex might be able to rescue tumour cells from a selective TORC1 blockade. For this reason, some novel double TORC1/2 inhibitors are in clinical development. A molecular relationship between mTOR and HNSCC has been defined (Freudlsperger et al, 2011), but the precise role of TORC2 in the onset and maintenance of resistance to EGFR blockade remains to be defined. Data from literature suggest that a more robust activity of novel dual PI3K/mTOR inhibitors, compared with either PI3K or mTOR inhibitors, is required for efficient suppression of tumour growth when multiple alterations of the PI3K pathway exist (Sabbah et al, 2011). On the basis of this body of evidence, the major purpose of our study is to analyse the role of PI3K/mTOR inhibition in HNSCC in the context of innate and acquired resistance to cetuximab, thus investigating the molecular mechanisms that might be involved in the development of resistance. To this aim, we tested the combination of cetuximab and PKI-587 on models of human squamous cancer cells with different degrees of sensitivity to cetuximab, evaluating their activity both in vitro and in vivo, in HNSCC tumour xenografts.
Materials and Methods
Compounds
Cetuximab was provided by ImClone Systems (Somerville, NJ, USA); PKI-587 (PF-05212384) was provided by Pfizer Inc (New York, NY, USA).
Cell cultures
Human squamous cancer cell lines A431 (epidermoid carcinoma), FaDu (pharyngeal carcinoma), Detroit562 (pharyngeal carcinoma) and Kyse30 (oesophageal carcinoma) were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) or Deutsche Sammlung von Mikroorganismen und Zellkulturen (DMSZ, Braunschweig, Germany). All cells were maintained according to the manufacturer’s protocol. A431-CR and FaDu-CR (cetuximab resistant) cells were generated through a validated protocol of in vivo/in vitro selection following chronic exposure to the drug, as recently described (Rosa et al, 2013); briefly, cells were injected s.c. in nude mice; when tumours reached ∼0.5 cm3, mice were treated with cetuximab 20 mg kg−1 i.p., three times a week for 8 weeks. Progressing tumours under treatment with cetuximab were excised and resistant cells were maintained in vitro and tested for sensitivity against the drug.
Cell density assay
Cells (104 cells per well) were grown in 24-well plates and exposed to increasing doses of cetuximab or PKI-587, alone or in combination. The percentage of cell density was determined using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay according to the manufacturer’s instructions. The dose–response curves for each agent alone and in combination were determined at a fixed ratio based on their IC50 values. IC50 was defined as the drug concentration causing 50% inhibition of cell proliferation.
Synergy analysis
The interaction between cetuximab and PKI-587 was determined using median-effect analysis, with the CalcuSyn ver. 2.0 software (Biosoft, Cambridge, UK), which calculates a non-exclusive case combination index (CI) for every fraction affected, a measure of the drug interaction effects. Combination index values of <1 or >1 indicated synergy and antagonism, respectively, whereas a CI value of 1 indicated additive effects of the drugs.
Western blot analysis
Total protein extracts obtained from cell cultures or tumour specimens were resolved by 4–15% SDS-PAGE and probed with anti-human, polyclonal pEGFR, polyclonal EGFR, monoclonal pMAPK, monoclonal MAPK, monoclonal beclin-1 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), polyclonal pAkt, polyclonal Akt, SQSTM1/p62 (Cell Signaling Technologies, Beverly, MA, USA), monoclonal pp70S6K and p70S6K (Upstate, Billerica, MA, USA) and monoclonal actin (Sigma-Aldrich, Milan, Italy). Immunoreactive proteins were visualised by enhanced chemiluminescence (Pierce, Rockford, IL, USA). Densitometry was performed by using Image J software (NIH, Bethesda, MD, USA).
Detection of caspase 3 activity
After treatment, cells were resuspended in a cell lysis buffer, and subjected to three freeze–thaw cycles at −70 °C. Caspase 3 activity was analysed using a Caspase-3 Colorimetric Assay Kit (CaspACE Assay System Colorimetric, Promega, Madison, WI, USA) according to the manufacturer’s protocol. The activity was measured with a microplate reader at 405 nm. Caspase specific activity was calculated as described in the manufacturer’s protocol.
Autophagy ELISA assay
After treatment, cells were resuspended in a RIPA cell lysis buffer as described in the manufacturer’s protocol. p62 levels in all cell lines were determined by ELISA (p62 ELISA kit, Enzo Life Science, Vinci, Italy). The absorbance was measured at 450 nm on a microplate reader.
Nude mouse cancer xenograft models
Five-week-old Balb/cAnNCrlBR athymic (nu+/nu+) mice (Charles River Laboratories, Milan, Italy) maintained in accordance with institutional guidelines of the University of Naples Animal Care Committee and in accordance with the Declaration of Helsinki were injected s.c. with Kyse30 human HNSCC cells (107 cells per mice) and resuspended in 200 μl of Matrigel (Collaborative Biomedical Products, Bedford, MA, USA). Seven days after the tumour cell injection, tumour-bearing mice were randomly assigned (n=10 per group) to receive the following: cetuximab 20 mg kg−1 i.p., three times a week for 3 weeks; PKI-587 25 mg kg−1 i.v., one time a week for 3 weeks (Mallon et al, 2011); or a combination of these agents. Tumour diameter was assessed with a vernier caliper, and tumour volume (cm3) was measured using the formula π/6 × larger diameter × (smaller diameter)2. Mice were killed when the tumour reached a size of ∼2 cm3, the maximum size allowed by the ethics committee.
Statistical analysis
Student’s t-test was used to evaluate the statistical significance of the in vitro results. The statistical significance of differences in tumour growth was determined by one-way ANOVA and Dunnett’s multiple comparison post-test, and that of differences in survival was determined by the log-rank test (Rosa et al, 2011). Linear regression analysis to determine the correlation between the IC50 of PKI-587 and the reduction of p62 and pro-caspase 3 levels was performed by using Sigma Plot ver. 11.0 (Systat Software, San Jose, CA, USA). All reported P-values were two-sided. All analyses were performed using the BMDP New System statistical package version 1.0 for Microsoft Windows (BMDP Statistical Software, Los Angeles, CA, USA).
Results
Inhibition of the PI3K-mTOR pathway is effective in HNSCC cell lines
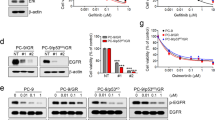
We selected human squamous cell cancer models with different sensitivities to the anti-EGFR mAb cetuximab, including A431 and FaDu cells, described as sensitive (Tijink et al, 2006; Meira et al, 2011), and Detroit562 and Kyse30 cells, described as poorly sensitive to EGFR inhibitors (Jimeno et al, 2007). According to the Sanger Institute catalogue of somatic mutations in cancer (*COSMIC database, Catalogue of Somatic Mutations In Cancer, http://www.sanger.ac.uk/), the mutational profile of the above cited cell lines is reported: Detroit562 cells harbour a H1047R mutation in the PI3K gene; Kyse30 cells harbour a Q61L mutation in the H-Ras gene. We also generated A431-CR and FaDu-CR cell lines with acquired resistance to cetuximab, through in vivo continuous exposition to the drug followed by tumour explant and in vitro stabilisation of the derived resistant cancer cell line, in agreement with the protocol we previously described (Bianco et al, 2008). We first analysed the in vitro sensitivity of all cell lines to cetuximab and to the dual PI3K/mTOR kinase inhibitor, PKI-587, through proliferation assay. The IC50 value was measured as the drug concentration causing 50% inhibition of cell proliferation. A431 and FaDu cells were sensitive to cetuximab exhibiting an IC50 value ⩽100 μg ml−1. However, Detroi562 and Kyse30 cells were resistant to cetuximab, exhibiting an IC50 value ⩾500 μg ml−1 (Figure 1A). All the human squamous cell lines were highly sensitive to PKI-587, with an IC50 value ⩽100 nM (Figure 1B).

Effects of cetuximab and PKI-587 on survival of HNSCC cell lines. Percent of cell density of human squamous cancer cells treated with increasing doses of cetuximab (20–500 μg ml−1) (A) and PKI-587 (10–5000 nM) (B), as measured by MTT assay. Data represent the mean (±s.d.) of three independent experiments, each performed in triplicate. Error bars indicate s.d. values.
PKI-587 treatment restores sensitivity to cetuximab in resistant HNSCC cell lines
The effect of cetuximab on cell growth and survival, alone and in combination with the dual PI3K/mTOR inhibitor, PKI-587, in both sensitive and resistant cells was evaluated. To better study the inhibitory effect and the possible synergism of PKI-587 and cetuximab co-treatment, we tested the combination of the two drugs at their equipotent ratio as described by Chou and Talalay, as shown in Figure 2 (Chou and Talalay, 1984). We measured the CIaccording to the Chou and Talalay method, using an automated calculation software. Based on this mathematical model, the combination is synergistic when CI is <1.0 and highly synergistic when CI is <0.5 (Supplementary Table S1). We found a highly synergistic effect in almost all tested combinations of PKI-587 and cetuximab across all cell lines (Figure 2A). Moreover, the addition of a very low single dose of PKI-587 (0.5 nM) with increasing doses of cetuximab enhances sensitivity to the latter in both resistant Detroit562 and Kyse30 cells (Supplementary Figure S1). These effects correlate with alterations on the expression/activation of several transducers involved in cell proliferation: whereas single treatment with cetuximab decreases phospho-Akt levels only in sensitive cells, PKI-587 is able to reduce it in all cell lines tested, but even more when used in combination with cetuximab (Figure 2B). Moreover, in all cell lines, sensitive or resistant to cetuximab, PKI-587 alone or combined with cetuximab induces the dephosphorylation/inactivation of the 70 kDa ribosomal protein S6 kinase (p70S6K), as confirmed through densitometric analysis; the effect on the RAS/MAPK pathway, as measured by extracellular-signal-regulated kinases 1/2 (ERK1/2) phosphorylation, seems marginal among cell lines, even if a decrease is observed upon combined treatment (Figure 2B).

Synergistic effect of cetuximab and PKI-587 in HNSCC cell lines. (A) Percent of cell density of human squamous cancer cells treated with cetuximab and PKI-587 at the equipotent ratio (IC30, IC40, IC50) as measured by MTT assay. Results represent the mean (±s.d.) of three independent experiments, each performed in triplicate. Error bars indicate s.d. values. Asterisks indicate statistical significance of combined treatment vs single agent, as determined by Student’s t-test (*, two-sided P<0.05; **, two-sided P<0.005). (B) Western blot analysis of protein expression in HNSCC cells treated for 24 h with cetuximab (200 μg ml−1), PKI-587 (50 nM) or their combination. Relative optical density of phospho-protein levels normalised to the total protein level were shown. Data represent the mean (±s.d.) of three independent experiments, each performed in triplicate, and are presented relative to control.
Caspase-dependent apoptosis is primarily activated in cetuximab sensitive cells
The described effect on cell survival signalling prompted us to investigate the putative role of different mechanisms of cell death in the context of cetuximab resistance. To this purpose, we analysed the effects of single-drug and combination treatments on the expression of some apoptotic/autophagy markers such as caspase 3, caspase 9, beclin-1 and p62. PKI-587 in combination with cetuximab is able to induce cell death in either sensitive or resistant cells; whereas combined treatment induces caspase activation only in sensitive cell lines, as measured by decreases in procaspase 3 and 9 levels, increased expression of beclin-1, paired with decrease in p62 levels, is detected only in resistant cell lines as confirmed through densitometry (Figure 3A). These data were confirmed by measuring caspase 3 activity and p62 levels through colorimetric and ELISA assays, respectively (Figures 3B and C). In A431 and FaDu cells, caspase 3 activity is significantly induced by PKI-587 treatment and even more by the addition of cetuximab (P-value <0.05, combination vs PKI-587) (Figure 3B), suggesting that activation of apoptosis is predominant in sensitive compared with Detroit562- and Kyse30-resistant HNSCC cells. By contrast, autophagy seems to be preferentially induced in resistant compared with sensitive cells, as shown by the decrease of p62 levels observed in Detroit562 and Kyse30 cells (Figure 3C), even if the addition of cetuximab does not potentiate this effect.

Effects of cetuximab and PKI-587 combination on apoptosis of HNSCC cell lines sensitive and resistant to cetuximab. (A) Western blot analysis of protein expression in human squamous cancer cells treated for 24 h with cetuximab (200 μg ml−l), PKI-587 (50 nM) or their combination. Relative optical density of protein levels normalised to the actin level were shown. Data represent the mean (±s.d.) of three independent experiments, each performed in triplicate, and are presented relative to control. (B) Activity of caspase 3 was measured by using a caspase 3 colorimetric protease assay kit. Values represent means±s.d. from triplicate samples for each treatment. Asterisks indicate statistical significance of combined treatment vs single agent, as determined by Student’s t-test (**, two-sided P<0.005). Error bars indicate s.d. values. (C) ELISA assay of p62 levels in human squamous cancer cells treated for 24 h with cetuximab (200 μg ml−1), PKI-587 (50 nM) or their combination. Values represent mean±s.d.values from triplicate samples for each treatment. Error bars indicate s.d.values.
Combination of cetuximab plus PKI-587 is effective in the condition of acquired resistance to cetuximab
To explore the role of the PI3K/Akt/mTOR pathway dependence in the context of resistance against EGFR blockade, we generated A431-CR and FaDu-CR cell lines with acquired resistance to cetuximab, through in vivo continuous exposition to the drug followed by tumour explant and in vitro stabilisation of the derived resistant cancer cell line (Bianco et al, 2008). We first analysed the in vitro sensitivity to cetuximab in cell lines with acquired resistance (A431-CR and FaDu-CR) compared with parental cell lines (Figure 4A). A431-CR and FaDu-CR cells appear to be resistant to cetuximab, exhibiting an IC50 value ⩾500 μg ml−1, similar to Detroit562 and Kyse30 cells. In the CR derivative cell lines, the effect of the dual PI3K/mTOR inhibitor, PKI-587, has been evaluated on cell growth and survival. As shown in Figure 4B, the combination treatments of PKI-587 and cetuximab are able to partially restore sensitivity to cetuximab even in presence of acquired resistance to the anti-EGFR mAb. The effects of single and combination treatments of cetuximab with PKI-587 on the expression/activation of several transducers involved in cell proliferation, such as Akt, p70S6K, MAPK, were then evaluated through western blot analysis. In cell lines with acquired resistance, the use of cetuximab does not induce alterations of the major downstream transducers of EGFR (Figure 4C). PKI-587 alone, but even more the combined treatment with cetuximab, induces the dephosphorylation/inactivation of Akt and p70S6K, as confirmed through densitometric analysis. Similar to intrinsically resistant cells,A431-CR and FaDu-CR cell lines also do not display pro-caspase modulation. Conversely, an increased expression of beclin-1, a major transducer of autophagy cell death, was observed after the combination treatment and confirmed through densitometric analysis (Figure 4C). The induction of autophagy is also demonstrated by the decrease of p62 levels measured through both western blot and ELISA assay (Figures 4C and D). In order to further evaluate the relationship between PKI-587 treatment and the described alterations, we analysed the correlation between the IC50 of PKI-587 and the protein reduction of both p62 and caspase 3 levels, in all CR cell lines (Detroit562, Kyse30, A431-CR and FaDu-CR) as described by Chang and Wang (2013)). As depicted in Supplementary Figure S2, there was a significant correlation between the IC50 of PKI-587 and the reduction of p62 protein levels (P<0.01) but not of caspase 3 levels in all four resistant cells (Supplementary Figure S2).

Effects of cetuximab and PKI-587 combination on survival of cells with acquired resistance to cetuximab. (A) Percent of cell density of human squamous cancer cells with acquired resistance to cetuximab (A431-CR, FaDu-CR) treated with increasing doses of cetuximab (20–500 μg ml−1) compared with parental cells (A431, FaDu). Data represent the mean (±s.d.) of three independent experiments, each performed in triplicate. Error bars indicate s.d. values. (B) Percent of cell density of cetuximab-resistant cells (A431-CR, FaDu-CR) treated with increasing doses of cetuximab (20–100 μg ml−1) and combination with PKI-587 (0.5 nM), as measured by MTT assay. Data represent the mean (±s.d.) of three independent experiments, each performed in triplicate. Error bars indicate s.d. values. Asterisks indicate statistical significance of combined treatment vs single agent, as determined by Student’s t-test (*, two-sided P<0.05; **, two-sided P<0.005). (C) Western blot analysis of protein expression in cetuximab-resistant cells treated for 24 h with cetuximab (200 μg ml−1), PKI-587 (50 nM) or their combination. Relative optical densities of phospho-protein levels or protein levels normalised to the total protein or actin level, respectively, are shown. Data represent the mean (±s.d.) of three independent experiments, each performed in triplicate, and are presented relative to control. (D) ELISA assay of p62 levels in cetuximab-resistant cells treated for 24 h with cetuximab (200 μg ml−1), PKI-587 (50 nM) or their combination. Values represent mean±s.d. from triplicate samples for each treatment. Error bars indicate s.d. values.
Cetuximab in combination with PKI-587 causes a cooperative antitumour effect in resistant HNSCC tumour xenografts
The antitumour effects observed in in vitro models of cetuximab resistance by the simultaneous blockade of EGFR and PI3K/mTOR prompted us to investigate such combination also in vivo. Balb/C nude mice xenografted with resistant Kyse30 tumours were treated with cetuximab or PKI-587, alone or in combination (Figure 5). Untreated mice xenografted with Kyse30 cells reached the maximum allowed tumour size of ∼2 cm3 on day 63, 9 weeks after tumour injection. At this time point, cetuximab or PKI-587 alone produced a growth inhibition of 40% and 70%, respectively, whereas the combined treatment induced a more pronounced inhibition of tumour growth (Figure 5A). Cetuximab-treated mice reached the tumour size of 2 cm3 on day 84, 8 weeks after treatment withdrawal, whereas mice treated with PKI-587 did not reach this size until the end of experiment, on day 98. PKI-587 alone showed strong antitumour activity, with 50% of growth inhibition until the end of the experiment. The combined treatment with cetuximab and PKI-587 persistently inhibited tumour growth, with an average tumour size of 0.3 cm3 until the end of the experiment, corresponding to a growth inhibition of >70%. Comparison of tumour sizes between the control group and combined treatment group, evaluated by the one-way ANOVA test, was statistically significant (P<0.001 at median survival of the control group) (Figure 5A). Furthermore, 40% of mice treated with the combination of cetuximab and PKI-587 were still alive at the end of the experiment. Accordingly, mice treated with the combination treatment showed a statistically significantly prolonged median survival compared with that of control mice (combination vs control, median survival 92 vs 44.5 days, hazard ratio=0.06621, 95% CI=0.01797–0.2439, P<0.0001). (Figure 5B). We then studied the effect of treatments on the expression of proteins having a critical role in cancer cell proliferation/survival. Western blot analysis was performed on lysates from tumours removed at the end of the third week of treatment, on day 31. As shown in Figure 5C, the combination treatment of cetuximab and PKI-587 induced the dephosphorylation/inactivation of Akt, p70S6K and MAPK; as also demonstrated in vitro, no evidence of caspase-dependent activation was found. Conversely, the combination treatment induced autophagy, as demonstrated by increase and decrease of the autophagy transducers beclin-1 and p62, respectively. These results were confirmed through densitometric analysis (Figure 5C).

Effect of cetuximab and PKI-587 combination on Kyse30 HNSCC tumour xenografts in nude mice. After 7 days from s.c. injection of Kyse30 cells, mice were randomised (10 per group) to receive cetuximab, PKI-587 or their combination, as described in Materials and Methods. The one-way ANOVA test was used to compare tumour sizes among different treatment groups at the median survival time of the control group (42 days). (A) They were statistically significant for cetuximab, PKI-587 and the combination vs control (P<0,005). Error bars indicate s.d. values. (B) Median survival was statistically significant for cetuximab, PKI-587 and their combination vs control (log-rank test). (C) Western blot analysis was performed on total lysates from Kyse30 tumour specimens of two mice treated as described in the methods section and killed on day 31. Relative optical density of phospho-protein levels or protein levels normalised to the total protein or actin level, respectively, are shown. Data represent the mean (±s.d.) of three independent experiments, each performed in triplicate, and are presented relative to control.
Discussion
The clinical use of cetuximab in the treatment of HNSCC is significantly limited by the intrinsic resistance observed in most patients and by the onset of acquired resistance arising in responders, despite its proven efficacy. Among several proposed mechanisms of resistance against EGFR inhibitors, the aberrant activation of intracellular signalling transducers appears to be of interest. For example, the activation of Akt/mTOR has been reported in 90% of HNSCC, independently from EGFR activation, by tumour array analysis (Molinolo et al, 2007). Moreover, aberrant expression of some key proteins, such as Akt, PTEN and mTOR, has been reported in HNSCC clinical tumour samples (Amornphimoltham et al, 2004; Nathan et al, 2007). Clinical trials are now investigating the activity of some PI3K inhibitors for incurable recurrent or metastatic HNSCC, including PX-866 (NCT01252628), BKM120 (NCT01816984) or BYL719 (NCT01602315), in combination with cetuximab. Although the overactivation of the PI3K pathway could contribute to HNSCC progression (Lui et al, 2013), little is known about the functional correlation of such alteration with resistance to cetuximab. The present study aimed to investigate the role of PI3K-dependent pathways in the context of innate and acquired resistance to anti-EGFR therapy; therefore, we selected human squamous cancer cellular models with different sensitivities to EGFR inhibitors, including A431 and FaDu cells, sensitive to cetuximab, Detroit562 and Kyse30 cells, with intrinsic resistance to cetuximab, and generated A431-CR and FaDu-CR cells, with acquired resistance to cetuximab. We hypothesised that pharmacological inhibition of the PI3K/Akt/mTOR pathway could revert the resistant phenotype. To this purpose, we analysed the effect of the dual PI3K/mTOR inhibitor, PF-05212384 (PKI-587), alone and in combination with cetuximab, in both sensitive and resistant cells.
Our data demonstrate that the treatment with PKI-587 is able to enhance sensitivity to cetuximab in HNSCC cells in vitro and to revert constitutive or acquired resistance. Interestingly, a major effect of the combined treatment is observed in Detroit562 cells, which harbour a PI3K mutation (H1047R substitution). These data are consistent with other reports showing the activity of dual inhibitors in other cancer types, including those with PI3K or K-RAS mutations. In CAL33 HNSCC cells, which harbour a PIK3CA mutation, the addition of the PI3K inhibitor LY294002 increases cetuximab sensitivity (Rebucci et al, 2011). In patient-derived xenografts of colorectal carcinomas, the triple therapy with NVP-BEZ235, a dual inhibitor of PI3K and mTOR, cetuximab and a MEK inhibitor provided improved disease control rate over single-agent therapies (Migliardi et al, 2012), although greater toxicity has been reported in preliminary clinical trials when the combination of dual PI3K/mTOR and MEK inhibitors was used (Shimizu et al, 2012). In EGFR-resistant non-small cell lung cancer cell lines, treatment with NVP-BEZ235 is able to revert the HGF-dependent resistant phenotype (Sano et al, 2013). Our data demonstrated that PKI-587 cells display improved antitumour activity when combined with cetuximab in resistant Kyse30 xenografts, with a prolonged advantage in terms of survival, compared with single-agent therapies. These effects were paired with an almost total suppression of the active phosphorylated form of Akt, reduction in phospo-p706Sk and phospho-ERK1/2 in tumour samples. These results are in line with those observed by Yang et al (2013) in nude mice xenografted with human nasopharyngeal carcinoma cells, in which a potent antitumour effect is reported when cisplatin is combined with NVP-BEZ235. It is known that PI3K activation could induce both pro-apoptotic or antiapoptotic effects (Castedo et al, 2002). Activated p70S6K cells, the major downstream effector of mTOR, as well as pAkt, are able to bind mitochondrial membranes and phosphorylate different pro-apoptotic molecules, which become inactive, thus promoting cell survival (Li et al, 2002). Conversely, mTOR is also able to translocate into the nucleus, resulting in phosphorylation and subsequent activation of p53.
This, in turn, induces the transcription of different pro-apoptotic proteins, finally resulting in caspase activation. Moreover, the activated form of Akt is able to directly phosphorylate and inhibit caspase 9 (Cardone et al, 1998). In addition, mTOR has been found to have profound effects on the regulation of cell death and control of apoptosis, a function that depends on the cellular context (Castedo et al, 2002). Therefore, we investigated the effect of double PI3K/mTOR inhibition on apoptosis signaling in the context of cetuximab resistance. Our data demonstrated that when PKI-587 is used in combination with cetuximab, despite the inhibition of proliferation and induction of cell death that are observed in either cetuximab-sensitive or -resistant cells, a caspase-dependent activation of apoptosis is detected primarily in sensitive cell lines. Therefore, we investigated alternative mechanisms of cell death or survival in response to PI3K/mTOR blockade in resistant cells, such as autophagy. Autophagy is a highly conserved catabolic cellular survival mechanism that allows for protein turnover in case of nutrient depletion and starvation (Sarbassov et al, 2005), cellular events that are in turn regulated by mTOR. However, although autophagy is best characterised as a survival response, it can also function as a mechanism of programmed cell death (Azad et al, 2008).
Consistently, in human glioma the treatment with NVP-BEZ235 induces autophagy (Liu et al, 2009), an effect observed also in hepatocellular carcinoma cells (Thomas et al, 2012). Regulation of beclin-1 appears to be an important mechanism to control autophagy. Several studies demonstrated that beclin-1 upregulation correlates with an increase in autophagy (Matsui et al, 2007; Pacheco et al, 2007). On the other hand, decreased p62 levels can be observed when autophagy is induced (Bjørkøy et al, 2009). On the basis of our data, a different balance between cell death by apoptosis and autophagy may be thought to exist in cetuximab-sensitive vs -resistant HNSCC cells. In fact, we found that only in cetuximab resistant cells an increase of beclin-1 and simultaneous reduction of p62 are observed upon combined treatment. Taken together, our results demonstrate that PKI-587 could act in HNSCC cells by inducing a different type of cell death. Furthermore, targeting the PI3K/mTOR pathways could restore sensitivity to the mAb cetuximab in the condition of both constitutive or acquired resistance to such agent.
Change history
10 June 2014
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Amornphimoltham P, Sriuranpong V, Patel V, Benavides F, Conti CJ, Sauk J, Sausville EA, Molinolo AA, Gutkind JS (2004) Persistent activation of the Akt pathway in head and neck squamous cell carcinoma: a potential target for UCN-01. Clin Cancer Res 10: 4029–4037.
Azad MB, Chen Y, Henson ES, Cizeau J, McMillan-Ward E, Israels SJ, Gibson SB (2008) Hypoxia induces autophagic cell death in apoptosis-competent cells through a mechanism involving BNIP3. Autophagy 4 (2): 195–204.
Baba Y, Fujii M, Tokumaru Y, Kato Y (2012) Present and future of EGFR inhibitors for head and neck squamous cell cancer. JOncol 2012: 986725.
Bancroft CC, Chen Z, Yeh J, Sunwoo JB, Yeh NT, Jackson S, Jackson C, Van Waes C (2002) Effects of pharmacologic antagonists of epidermal growth factor receptor, PI3K and MEK signal kinases on NF-kappaB and AP-1 activation and IL-8 and VEGF expression in human head and neck squamous cell carcinoma lines. Int J Cancer 99 (4): 538–548.
Bianco R, Daniele G, Ciardiello F, Tortora G (2005) Monoclonal antibodies targeting the epidermal growth factor receptor. Curr Drug Targets 6 (3): 275–287.
Bianco R, Garofalo S, Rosa R, Damiano V, Gelardi T, Daniele G, Marciano R, Ciardiello F, Tortora G (2008) Inhibition of mTOR pathway by everolimus cooperates with EGFR inhibitors in human tumours sensitive and resistant to anti-EGFR drugs. Br J Cancer 98: 923–930.
Bianco R, Rosa R, Damiano V, Daniele G, Gelardi T, Garofalo S, Tarallo V, De Falco S, Melisi D, Benelli R, Albini A, Ryan A, Ciardiello F, Tortora G (2008) Vascular endothelial growth factor receptor-1 contributes to resistance to anti-epidermal growth factor receptor drugs in human cancer cells. Clin Cancer Res 14 (16): 5069–5080.
Bjørkøy G, Lamark T, Pankiv S, Øvervatn A, Brech A, Johansen T (2009) Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol 452: 181–197.
Cardone MH, Roy N, Stennicke HR, Salvesen GS, Franke TF, Stanbridge E, Frisch S, Reed JC (1998) Regulation of cell death protease caspase-9 by phosphorylation. Science 282 (5392): 1318–1321.
Castedo M, Ferri KF, Kroeme G (2002) Mammalian target of rapamycin(mTOR):pro-and anti-apoptotic. Cell Death Differ 9 (2): 99–100.
Chang AY, Wang M (2013) Molecular mechanisms of action and potential biomarkers of growth inhibition of dasatinib (BMS-354825) on hepatocellular carcinoma cells. BMC Cancer 13 (1): 267.
Chou TC, Talalay P (1984) Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 22: 27–55.
Engelman JA, Luo J, Cantley LC (2006) The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet 7: 606–619.
Freudlsperger C, Burnett JR, Friedman JA, Kannabiran VR, Chen Z, Van Waes C (2011) EGFRPI3K- AKT-mTOR signaling in head and neck squamous cell carcinomas: attractive targets for molecular-oriented therapy. Expert Opin Ther Targets 15 (1): 63–74.
Guertin DA, Sabatini DM (2007) Defining the role of mTOR in cancer. Cancer Cell 12 (1): 9–22.
Herzog A, Bian Y, Vander Broek R, Hall B, Coupar J, Cheng H, Sowers AL, Cook JD, Mitchell JB, Chen Z, Kulkarni AB, Van Waes C (2013) PI3K/mTOR inhibitor PF-04691502 antitumor activity is enhanced with induction of wild-type TP53 in human xenograft and murine knockout models of head and neck cancer. Clin Cancer Res 19 (14): 3808–3819.
Jimeno A, Kulesza P, Wheelhouse J, Chan A, Zhang X, Kincaid E, Chen R, Clark DP, Forastiere A, Hidalgo M (2007) Dual EGFR and mTOR targeting in squamous cell carcinoma models, and development of early markers of efficacy. Br J Cancer 96 (6): 952–959.
Li B, Desai S, MacCorkle-Chosenek R, Fan L, Spencer D (2002) A novel conditional AKT ‘survival switch’ reversibly protects cells from apoptosis. Gene Ther 9 (4): 233–244.
Liu TJ, Koul D, LaFortune T, Tiao N, Shen RJ, Maira SM, Garcia-Echevrria C, Yung WK (2009) NVP-BEZ235, a novel dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor, elicits multifaceted antitumor activities in human gliomas. Mol Cancer Ther 8 (8): 2204–2210.
Lui VW, Hedberg ML, Li H, Vangara BS, Pendleton K, Zeng Y, Lu Y, Zhang Q, Du Y, Gilbert BR, Freilino M, Sauerwein S, Peyser ND, Xiao D, Diergaarde B, Wang L, Chiosea S, Seethala R, Johnson JT, Kim S, Duvvuri U, Ferris RL, Romkes M, Nukui T, Kwok-Shing NgP, Garraway LA, Hammerman PS, Mills GB, Grandis JR (2013) Frequent mutation of the PI3K pathway in head and neck cancer defines predictive biomarkers. Cancer Discov 3 (7): 761–769.
Mallon R, Feldberg LR, Lucas J, Chaudhary I, Dehnhardt C, Santos ED, Chen Z, dos Santos O, Ayral-Kaloustian S, Venkatesan A, Hollander I (2011) Antitumor efficacy of PKI-587, a highly potent dual PI3K/mTOR kinase inhibitor. Clin Cancer Res 17 (10): 3193–3203.
Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J (2007) Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMPactivated protein kinase and Beclin 1 in mediating autophagy. Circ Res 100 (6): 914–922.
Maurizi M, Almadori G, Ferrandina G, Distefano M, Romanini ME, Cadoni G, Benedetti Panici P, Paludetti G, Scambia G, Mancuso S (1996) Prognostic significance of epidermal growth factor receptor in laryngeal squamous cell carcinoma. Br J Cancer 74 (8): 1253–1257.
Meira DD, Almeida VH, Mororó JS, Caetano MS, Nóbrega IP, Batista D, Sternberg C, Ferreira CG (2011) Efficient blockade of Akt signaling is a determinant factor to overcome resistance tomatuzumab. Mol Cancer 10: 151.
Migliardi G, Sassi F, Torti D, Galimi F, Zanella ER, Buscarino M, Ribero D, Muratore A, Massucco P, Pisacane A, Risio M, Capussotti L, Marsoni S, Di Nicolantonio F, Bardelli A, Comoglio PM, Trusolino L, Bertotti A (2012) Inhibition of MEK and PI3K/mTOR suppresses tumor growth but does not cause tumor regression in patient-derived xenografts of RAS-mutant colorectal carcinomas. Clin Cancer Res 18 (9): 2515–2525.
Molinolo AA, Hewitt SM, Amornphimoltham P, Keelawat S, Rangdaeng S, Meneses García A, Raimondi AR, Jufe R, Itoiz M, Gao Y, Saranath D, Kaleebi GS, Yoo GH, Leak L, Myers EM, Shintani S, Wong D, Massey HD, Yeudall WA, Lonardo F, Ensley J, Gutkind JS (2007) Dissecting the Akt/mammalian target of rapamycin signaling network: emerging results from the head and neck cancer tissue array initiative. Clin Cancer Res 13 (17): 4964–4973.
Moroni M, Veronese S, Benvenuti S, Marrapese G, Sartore-Bianchi A, Di Nicolantonio F, Gambacorta M, Siena S, Bardelli A (2005) Gene copy number for epidermal growth factor receptor (EGFR) and clinical response to antiEGFR treatment in colorectal cancer: a cohort study. Lancet Oncol 6: 279–286.
Nathan CO, Amirghahari N, Rong X, Giordano T, Sibley D, Nordberg M, Glass J, Agarwal A, Caldito G (2007) Mammalian target of rapamycin inhibitors as possible adjuvant therapy for microscopic residual disease in head and neck squamous cell cancer. Cancer Res 67: 2160–2168.
Nestor M (2010) Effect of cetuximab treatment in squamous cell carcinomas. Tumour Biol 31 (2): 141–147.
Pacheco CD, Kunkel R, Lieberman AP (2007) Autophagy in Niemann-Pick C disease is dependent upon Beclin-1 and responsive to lipid trafficking defects. Hum Mol Genet 16 (12): 1495–1503.
Pernas FG, Allen CT, Winters ME, Yan B, Friedman J, Dabir B, Saigal K, Mundinger GS, Xu X, Morris JC, Calvo KR, Van Waes C, Chen Z (2009) Proteomic signatures of epidermal growth factor receptor and survival signal pathways correspond to gefitinib sensitivity in head and neck cancer. Clin Cancer Res 15 (7): 2361–2372.
Pedrero JM, Carracedo DG, Pinto CM, Zapatero AH, Rodrigo JP, Nieto CS, Gonzalez MV (2005) Frequent genetic and biochemical alterations of the PI 3-K/AKT/PTEN pathway in head and neck squamous cell carcinoma. Int J Cancer 114 (2): 242–248.
Rebucci M, Peixoto P, Dewitte A, Wattez N, De Nuncques MA, Rezvoy N, Vautravers-Dewas C, Buisine MP, Guerin E, Peyrat JP, Lartigau E, Lansiaux A (2011) Mechanisms underlying resistance to cetuximab in the HNSCC cell line: role of AKT inhibition in bypassing this resistance. Int J Oncol 38 (1): 189–200.
Rosa R, Melisi D, Damiano V, Bianco R, Garofalo S, Gelardi T, Agrawal S, Di Nicolantonio F, Scarpa A, Bardelli A, Tortora G (2011) Toll-like receptor 9 agonist IMO cooperates with cetuximab in K-ras mutant colorectal and pancreatic cancers. Clin Cancer Res 17 (20): 6531–6541.
Rosa R, Monteleone F, Zambrano N, Bianco R (2013) In vitro and in vivo models for analysis of resistance to anticancer molecular therapies. Curr Med Chem 21 (14): 1595–1606.
Sabbah DA, Brattain MG, Zhong H (2011) Dual inhibitors of PI3K/mTOR or mTOR-selective inhibitors: which way shall we go? Curr Med Chem 18 (36): 5528–5544.
Sano T, Takeuchi S, Nakagawa T, Ishikawa D, Nanjo S, Yamada T, Nakamura T, Matsumoto K, Yano S (2013) The novel phosphoinositide 3-kinase-mammalian target of rapamycin inhibitor,BEZ235, circumvents erlotinib resistance of epidermal growth factor receptor mutant lung cancer cells triggered by hepatocyte growth factor. Int J Cancer 133 (2): 505–513.
Sarbassov DD, Ali SM, Sabatini DM (2005) Growing roles for the mTOR pathway. Curr Opin Cell Biol 17 (6): 596–603.
Shaw RJ, Cantley LC (2006) Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 441: 424–430.
Shimizu T, Tolcher AW, Papadopoulos KP, Beeram M, Rasco DW, Smith LS, Gunn S, Smetzer L, Mays TA, Kaiser B, Wick MJ, Alvarez C, Cavazos A, Mangold GL, Patnaik A (2012) The clinical effect of the dual-targeting strategy involving PI3K/AKT/mTOR and RAS/MEK/ERK pathways in patients with advanced cancer. Clin Cancer Res 18 (8): 2316–2325.
Thomas HE, Mercer CA, Carnevalli LS, Park J, Andersen JB, Conner EA, Tanaka K, Matsutani T, Iwanami A, Aronow BJ, Manway L, Maira SM, Thorgeirsson SS, Mischel PS, Thomas G, Kozma SC (2012) mTOR inhibitors synergize on regression, reversal of gene expression, and autophagy in hepatocellular carcinoma. Sci Transl Med 4 (139): 139ra84.
Tijink BM, Neri D, Leemans CR, Budde M, Dinkelborg LM, Stigter-van Walsum M, Zardi L, van Dongen GA (2006) Radioimmunotherapy of head and neck cancer xenografts using 131Ilabeled antibody L19-SIP for selective targeting of tumor vasculature. J Nucl Med 47 (7): 1127–1135.
Van Waes C, Allen CT, Citrin D, Gius D, Colevas AD, Harold NA, Rudy S, Nottingham L, Muir C, Chen Z, Singh AK, Dancey J, Morris JC (2010) Molecular and clinical responses in a pilot study of gefitinib with paclitaxel and radiation in locally advanced head-and-neck cancer. Int J Radiat Oncol Biol Phys 77 (2): 447–454.
Yang F, Qian XJ, Qin W, Deng R, Wu XQ, Qin J, Feng GK, Zhu XF (2013) Dual phosphoinositide 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235 has a therapeutic potential and sensitizes cisplatin in nasopharyngeal carcinoma. PLoS One 8 (3): e59879.
Yuan TL, Cantley LC (2008) PI3K pathway alterations in cancer: variations on a theme. Oncogene 27: 5497–5510.
Acknowledgements
This study was supported by Associazione Italiana per la Ricerca sul Cancro (AIRC) My First Grant 2011-2014 (MFAG-11473) to RB.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
D'Amato, V., Rosa, R., D'Amato, C. et al. The dual PI3K/mTOR inhibitor PKI-587 enhances sensitivity to cetuximab in EGFR-resistant human head and neck cancer models. Br J Cancer 110, 2887–2895 (2014). https://doi.org/10.1038/bjc.2014.241
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2014.241
Keywords
This article is cited by
-
Platycodin D represses β-catenin to suppress metastasis of cetuximab-treated KRAS wild-type colorectal cancer cells
Clinical & Experimental Metastasis (2023)
-
Phase I trial of copanlisib, a selective PI3K inhibitor, in combination with cetuximab in patients with recurrent and/or metastatic head and neck squamous cell carcinoma
Investigational New Drugs (2021)
-
A phospho-proteomic study of cetuximab resistance in KRAS/NRAS/BRAFV600 wild-type colorectal cancer
Cellular Oncology (2021)
-
Targeting mTOR for cancer therapy
Journal of Hematology & Oncology (2019)
-
Targeting phosphoinositide 3-kinase (PI3K) in head and neck squamous cell carcinoma (HNSCC)
Cancers of the Head & Neck (2018)

{kind=link}
{kind=link}
{kind=link}