
Abstract
Background:
The optimal interval between two consecutive mammograms is uncertain. The UK Frequency Trial did not show a significant difference in breast cancer mortality between screening every year (study group) and screening every 3 years (control group). In this study, the trial is simulated in order to gain insight into the results of the trial and to predict the effect of different screening intervals on breast cancer mortality.
Methods:
UK incidence, life tables and information from the trial were used in the microsimulation model MISCAN–Fadia to simulate the trial and predict the number of breast cancer deaths in each group. To be able to replicate the trial, a relatively low sensitivity had to be assumed.
Results:
The model simulated a larger difference in tumour size distribution between the two groups than observed and a relative risk (RR) of 0.83 of dying from breast cancer in the study group compared with the control group. The predicted RR is lower than that reported from the trial (RR 0.93), but within its 95% confidence interval (0.63–1.37).
Conclusion:
The present study suggests that there is benefit of shortening the screening interval, although the benefit is probably not large enough to start annual screening.
Similar content being viewed by others


Main
In randomised controlled trials, mammography screening has been shown to reduce breast cancer mortality rates (Tabar et al, 1992; Nystrom et al, 1993, 2002). The more frequently a woman has screening exams, the larger the probability of having an early diagnosis, and the larger the mortality reduction might be. However, with more frequent exams, the potential of false-positive exams and overdiagnosis will also increase (Jansen and Zoetelief, 1997; Christiansen et al, 2000). There is no consensus on the optimal screening interval (i.e., the time between two consecutive mammograms), as is illustrated by the variety of screening intervals used throughout the world. Most European screening programmes use an interval of 2 years (e.g., The Netherlands, Sweden), whereas other countries use a 3-year interval (United Kingdom, Malta). Even within the same country, screening recommendations vary: in the United States, the American Cancer Society recommends annual screening starting at the age of 40 years (Smith et al, 2010), whereas the US Preventive Services Task Force recently changed their recommendation to biennial screening from age 50 to 74 years (US Preventive Services Task Force, 2009).
Two randomised trials compared a 1-year screening interval with a 3-year screening interval, one in women between age 40 and 49 years (Klemi et al, 1997) and one in women between age 50 and 62 years (Breast Screening Frequency Trial Group, 2002). The latter, the UK Breast Screening Frequency Trial, was conducted from 1989 to 1996, in order to evaluate the difference in (predicted) breast cancer mortality between screening annually and screening once every 3 years (Breast Screening Frequency Trial Group, 2002). The tumours in the trial group, offered annual screening, were significantly smaller than those diagnosed in the control group, offered screening every 3 years. For node status and histological grade, no significant difference between the two groups was found. The initially reported relative risk (RR) predicted on the basis of two prognostic indices showed a nonsignificant reduction in predicted breast cancer mortality (Breast Screening Frequency Trial Group, 2002). The results were later updated with results on the actual observed number of breast cancer deaths in both groups again showing a nonsignificant reduction in breast cancer mortality. Women in the study group had an RR of 0.93 (95% confidence interval (CI) 0.63–1.37) of dying from breast cancer compared with women in the control group (Duffy and Blamey, 2008).
This finding was (slightly) surprising and raised the question why no significant difference was found between the two groups. It might be that there is truly only a very small mortality benefit of more frequent screening, or there might be other reasons why no difference is found between the two groups, for example, a lack of power or low sensitivity of mammography. Most policy predictions are based on the assumption that increasing the screening frequency will lead to more early diagnoses and consequently in a reduction in breast cancer mortality, hence it is crucial to get more insight in the results of this trial. A simulation model is ideally suited to evaluate the effect of different screening intervals on mortality, because the effect of different screening test sensitivities can be assessed and the model guarantees that trial populations are identical, except for the factors investigated.
In the present study, the UK Breast Screening Frequency Trial was simulated using the microsimulation model MIcrosimulation of SCreening ANalysis–Fatal diameter (MISCAN–Fadia), in order to gain insight into the results of the trial and estimate the effect of different screening intervals on breast cancer mortality.
Materials and methods
Model overview
MISCAN–Fadia is a microsimulation model developed within the Cancer Intervention and Surveillance Modeling Network (CISNET) (Tan et al, 2006). Briefly, the model simulates life histories in the absence of screening and then assesses how these life histories change as a consequence of screening programmes. MISCAN–Fadia explicitly models invasive tumour growth in combination with the concept of a fatal diameter. The model has been described in detail elsewhere (Tan et al, 2006) and information about the model can be found on the CISNET website (http://cisnet.cancer.gov/). A detailed description of the model components and model quantification for the present study is presented in the Appendix.
In brief, for the present study, the model simulates a population of women between the ages of 50 and 62 years in the year 1992 (i.e., the middle year of the trial) using the life tables of the UK female population. Among those who develop breast cancer, the natural history is modelled as a continuously growing tumour. Each tumour has a size (the fatal diameter, which differs between tumours) at which diagnosis and treatment will no longer result in cure given available treatment options. If the tumour is diagnosed (either on the basis of clinical presentation with symptoms or by screening) and treated before it has reached its fatal diameter, the woman will be cured and will die of non-breast cancer causes. Variation between tumours is modelled by probability distributions of tumour growth, threshold diameter of screen detection, clinical diagnosis diameter and fatal disease diameter.
When a screening programme is applied, the pre-clinical tumour may be detected by screening. Each simulated tumour has a diameter at which it will be clinically diagnosed as well as a screen-detection threshold diameter. For the latter, screening test sensitivity is 0% below and 100% above this diameter. The threshold diameter is assumed to decrease with age and calendar year. Screening benefits result from detection of more tumours at a non-fatal size (Tan et al, 2006).
Model calibration and validation
Several approaches have been used to assess the internal reliability of MISCAN–Fadia and the validity of the results against external data, as previously reported (Tan et al, 2006). For the present study, age-specific breast cancer incidence rates for the years 1975–1988, that is, before the implementation of the National Health Service Breast Screening Programme (NHSBSP) were used to estimate age-specific parameters for disease onset. The age-specific breast cancer incidence rates for the year 1988 as simulated by MISCAN–Fadia were compared with the observed incidence rates for the year 1988 in the United Kingdom.
UK breast screening frequency trial
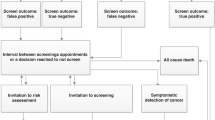
Five screening units participated in the trial held between 1989 and 1996 (Breast Screening Frequency Trial Group, 2002) (see Figure 1 for an overview of the trial design). A total of 99 389 women aged 50–62 years who had been invited to a prevalence screen in the NHSBSP were randomised to a conventional screen after an interval of 3 years (control group, n=50 216), or to three annual screenings (study group, n=49 173). For the primary analysis, only women who attended the prevalence screen and in whom no cancer was found at the prevalence screen were included (n=38 492 in the control group and n=37 530 in the study group). The attendance rate in the control group, among women who had attended the prevalence screen, was 85%. In the study group, attendance rates at the three yearly screens were 78%, 78% and 81%, respectively, (Breast Screening Frequency Trial Group, 2002).

A schematic overview of the UK Breast Screening Frequency Trial.
Trial replication and mortality prediction
Initially, the model based on data from randomised screening trials (extrapolated to the current period) and US data simulated a more favourable tumour size distribution than observed in the trial for both groups. Therefore, the threshold diameter and diameter of clinical detection were estimated using data from the Frequency Trial on the numbers of invasive breast cancers in both groups of the trial split out by tumour size and detection mode (see Appendix). Compared with the initially used values, the estimated values were somewhat higher for the diameter of clinical detection and the threshold diameter, corresponding to a lower screening sensitivity.
Subsequently, this fitted model was used to predict the number of breast cancer deaths from cancers diagnosed during the trial period in each group with a follow-up period up to 2006. From these numbers, a predicted RR of dying from breast cancer in the study group compared with the control group was calculated.
In addition, we investigated the effect of a longer follow-up period (i.e., until all women have died), a higher sensitivity (using the initial value for the threshold diameter) and full compliance (i.e., 100% attendance rates) on the predicted RR.
Results
Model calibration and validation
The observed age-specific incidence rates in the year 1988 as reported by the NHSBSP were accurately reproduced by MISCAN–Fadia (Figure 2). For each 5-year age group (35–79 years), the difference between the observed and simulated incidence rates was <10%.

Age-specific breast cancer incidence rates in the UK for the year 1988 as observed and simulated by MISCAN–Fadia.
Trial replication and mortality prediction
The model with the threshold diameter and diameter of clinical detection estimated based on the trial data, simulated a total of 523 (445 invasive) breast cancers in women who attended the prevalence screen compared with a total of 535 (443 invasive) cancers observed in the trial. The numbers of detected breast cancers and percentages screen detected, and clinically detected cancers are close to the observed numbers and percentages in both the groups (Table 1).
For the trial period, the cumulative incidence (number of invasive breast cancers detected) in both groups over time since prevalence screen, as observed in the trial and simulated by the model, is shown in Figure 3.

Cumulative incidence (number of invasive breast cancers) over time since prevalence screen in the control and study group as observed in the Frequency Trial (obs) and simulated by MISCAN–Fadia (sim).
The model simulated a more favourable tumour size distribution in the study group than in the control group, in line with what was observed in the trial (Table 2). For the control group, the simulated size distribution was somewhat less favourable (61% small tumours simulated vs 66% observed) and for the study group, the simulated size distribution was somewhat too favourable (77% small tumours simulated vs 73% observed). Thus, the model simulated a larger difference in size distribution between the control and study group than observed.
In the control group, 55 breast cancer deaths from cancers diagnosed in the trial were observed during the median follow-up of 162 months (Duffy and Blamey, 2008), compared with 54 deaths predicted. In the study group 50 breast cancer deaths were observed (Duffy and Blamey, 2008), whereas the model predicted 45 breast cancer deaths. The predicted difference between the number of deaths in the control group and the study group was larger than the observed difference, corresponding to a predicted RR of 0.83 of dying from breast cancer in the study group compared with the control group.
A longer follow-up period (life-time) had no effect on the predicted RR (Table 3). Increasing the sensitivity led to a higher percentage of screen-detected cancers in both groups (78% in the study and 58% in the control group) and to a lower predicted RR of 0.81, as did increasing the attendance to 100%. The combination of full compliance and higher sensitivity led to a predicted RR of 0.77.
Discussion
The present study suggests that there is benefit in terms of a reduction in breast cancer mortality associated with shortening the screening interval from 3 years to 1 year. The results show that if the available information from the UK Breast Screening Frequency Trial is used in a microsimulation model that is based on the results of randomised screening trials including a large(r) number of women, a larger effect of shortening the screening interval is predicted. The predicted RR of breast cancer death for the study group (offered three annual screens) compared with the control group (offered one screen after 3 years) was 0.83. The effect of more frequent screening is predicted to be larger when the attendance rate or screening test sensitivity is increased.
The microsimulation model used in the present study fitted the data better when a somewhat higher threshold diameter, corresponding to a lower screening test sensitivity, and diameter of clinical detection is used compared with a model based on data from randomised screening trials (extrapolated to the current period) and from the US. Thus, it seems that the screening test sensitivity in the trial was relatively low, which is in line with previously reported results showing that screen-detected as well as interval cancers could benefit from improved sensitivity (Warren et al, 2003). The results of the present study indicate that when the screening test sensitivity is higher, the effect of shortening the screening interval will be somewhat larger. This finding is important when considering shorter screening intervals for certain risk groups. For example, it has been hypothesised that women with high breast density might benefit more from additional frequent screening, because they have a higher risk of breast cancer (Kerlikowske et al, 2010). However, screening test sensitivity has been found to be lower in women with high breast density (Carney et al, 2003). The present study shows that the lower sensitivity in this group might offset some of the potential benefit of more frequent screening in this group.
The most important limitation of the current study is the relative paucity of data available to simulate the trial. More information (e.g., on the age distribution of participants and tumour size distribution of screen vs clinically detected cancers in both groups) might further improve the model, and consequently the model predictions. In addition, detailed information on the attendance rates was not available. For example, the non-attendance in the study group was somewhat higher than in the control group (approximately 20% vs 15%). It is unknown which proportion of the non-attenders in the study group missed multiple rounds (Andersson, 2002). Including more detailed information in the model will lead to better estimates of the effect of shortening the screening interval.
In addition, only one simulation model was used to estimate the effect of 1-year vs 3-year screening intervals. Having multiple models that come to similar findings might have strengthened the conclusion of the present study.
Despite these limitations, the microsimulation model, used in this study, adequately simulated the number of screen detected and interval cancers in both arms of the trial. Moreover, the predicted numbers of breast cancer deaths from cancers diagnosed in the trial were of the same magnitude as that of the reported numbers in both groups (Duffy and Blamey, 2008), and the predicted RR is within the 95% CI of the estimate reported from the trial.
The UK Breast Screening Frequency Trial showed a nonsignificant 7% reduction in breast cancer deaths in the study vs the control group (Duffy and Blamey, 2008), whereas the present study finds a substantially larger effect of shortening the screening interval (17%). The question arises why the model outcomes differ from the trial results. Several factors might contribute.
First, the RR predicted by the model is within the 95% CI of the trial-reported RR, indicating that the predicted RR is not statistically different from the trial-reported RR. The Frequency Trial invited 99 389 women, based on an expected 25% difference in breast cancer mortality between the study and control group (Day and Duffy, 1996). The current study shows that this estimated difference of 25% was too optimistic, suggesting that the trial was underpowered to find a significant difference between the two groups. On the basis of the results of the current study (i.e., an RR of 0.83), approximately 945 000 women needed to have been invited for a power of 80% to demonstrate a significant (P=0.05) difference in breast cancer mortality between the two groups (Lwanga and Lemeshow, 1991). However, the trial was designed to show a difference in predicted mortality, based on surrogate end points; in this case, the tumour size of the detected cancers. It was estimated that the sample size can be 2.74 times smaller without losing precision when surrogate end points are used (Day and Duffy, 1996). This means that when surrogate end points (such as prognostic indices) are used, at least 345 000 women needed to have been invited in order to have 80% power. Our findings indicate that the number needed to invite can also be reduced by increasing compliance to screening tests or increasing screening test sensitivity.
Furthermore, in the trial, more invasive breast cancers were detected in the study group than in the control group. Thus, more diagnoses have been moved forward in time in the study group than in the control group and then, more breast cancer deaths from cancers diagnosed in trial can be expected in the study group. An alternative would be to compare the number of breast cancer deaths from all breast cancers (during a certain follow-up period). However, after the trial, everyone receives usual care (triennial screening), resulting in a dilution of the effect on mortality. Both comparisons will lead to an underestimation of the effect of more frequent screening (i.e., a bias towards an RR of 1).
The results of the available observational studies are somewhat contradictory on the effect of shortening the screening interval. For example, two retrospective studies showed similar prognostic factors for women screened annually vs biennially (White et al, 2004; Wai et al, 2005). However, two other studies found that women who were screened annually had breast tumours that were smaller and less advanced than those who were screened every other year (Field et al, 1998; Hunt et al, 1999). Furthermore, six independent models showed that there is some benefit when moving from biennial to annual screening, although the benefit diminished (i.e., the benefit of moving from biennial to annual screening is smaller than that of moving from no screening to biennial screening). For example, 68–90% of the benefit is maintained when moving from annual to biennial screening scenarios for women aged 50–69 years (Mandelblatt et al, 2009). Thus, the benefit of screening every year is not three times as large as screening carried out once in every 3 years. The associated harms and costs also have to be taken into account when determining the optimal screening frequency and increases more steeply with more frequent screening than the benefits.
In conclusion, the present study suggests that there is benefit in terms of a mortality reduction of shortening the screening interval from 3 years to 1 year. However, the benefit is probably not large enough to start annual screening (Boer et al, 1998). At the same time, there seems to be no reason to abolish the 2-year interval currently used in most European screening programmes. For these programmes, benefits in terms of mortality reductions have been shown (Otto et al, 2003; Tabar et al, 2003).
Change history
29 March 2012
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Andersson I (2002) Comment on ‘The frequency of breast cancer screening: results from the UKCCCR Randomised Trial’. Eur J Cancer 38: 1427–1428; discussion 1465
Boer R, de Koning H, Threlfall A, Warmerdam P, Street A, Friedman E, Woodman C (1998) Cost effectiveness of shortening screening interval or extending age range of NHS breast screening programme: computer simulation study. BMJ 317: 376–379
Breast Screening Frequency Trial Group (2002) The frequency of breast cancer screening: results from the UKCCCR Randomised Trial United Kingdom Co-ordinating Committee on Cancer Research. Eur J Cancer 38: 1458–1464
Carney PA, Miglioretti DL, Yankaskas BC, Kerlikowske K, Rosenberg R, Rutter CM, Geller BM, Abraham LA, Taplin SH, Dignan M, Cutter G, Ballard-Barbash R (2003) Individual and combined effects of age, breast density, and hormone replacement therapy use on the accuracy of screening mammography. Ann Intern Med 138: 168–175
Christiansen CL, Wang F, Barton MB, Kreuter W, Elmore JG, Gelfand AE, Fletcher SW (2000) Predicting the cumulative risk of false-positive mammograms. J Natl Cancer Inst 92: 1657–1666
Day NE, Duffy SW (1996) Trial design based on surrogate end points – application to comparison of different breast screening frequencies. J R Statist Soc A 159: 49–60
Duffy SW, Blamey R (2008) Long-term Mortality Results From The UK Screening Frequency Trial In 6th European Breast Cancer Conference: Berlin, Germany
Early Breast Cancer Trialists’ Collaborative Group (1998a) Polychemotherapy for early breast cancer: an overview of the randomised trials. Lancet 352 (9132): 930–942
Early Breast Cancer Trialists’ Collaborative Group (1998b) Tamoxifen for early breast cancer: an overview of the randomised trials. Lancet 351 (9114): 1451–1467
Field LR, Wilson TE, Strawderman M, Gabriel H, Helvie MA (1998) Mammographic screening in women more than 64 years old: a comparison of 1- and 2-year intervals. AJR Am J Roentgenol 170: 961–965
Human Mortality Database University of California, Berkeley (USA), and Max Planck Institute for Demographic Research (Germany). Available from: http://www.mortality.org
Hunt KA, Rosen EL, Sickles EA (1999) Outcome analysis for women undergoing annual vs biennial screening mammography: a review of 24 211 examinations. AJR Am J Roentgenol 173: 285–289
Jansen JT, Zoetelief J (1997) Optimisation of mammographic breast cancer screening using a computer simulation model. Eur J Radiol 24: 137–144
Kerlikowske K, Cook AJ, Buist DS, Cummings SR, Vachon C, Vacek P, Miglioretti DL (2010) Breast cancer risk by breast density, menopause, and postmenopausal hormone therapy use. J Clin Oncol 28: 3830–3837
Klemi PJ, Toikkanen S, Rasanen O, Parvinen I, Joensuu H (1997) Mammography screening interval and the frequency of interval cancers in a population-based screening. Br J Cancer 75: 762–766
Lwanga SK, Lemeshow S (1991) Sample Size Determination In Health Studies: A Practical Manual. World Health Organization: Geneva
Mandelblatt JS, Cronin KA, Bailey S, Berry DA, de Koning HJ, Draisma G, Huang H, Lee SJ, Munsell M, Plevritis SK, Ravdin P, Schechter CB, Sigal B, Stoto MA, Stout NK, van Ravesteyn NT, Venier J, Zelen M, Feuer EJ, Breast Cancer Working Group of the Cancer Intervention and Surveillance Modeling Network (2009) Effects of mammography screening under different screening schedules: model estimates of potential benefits and harms. Ann Intern Med 151: 738–747
Mariotto AB, Feuer EJ, Harlan LC, Abrams J (2006) Dissemination of adjuvant multiagent chemotherapy and tamoxifen for breast cancer in the United States using oestrogen receptor information: 1975–1999. J Natl Cancer Inst Monogr 2006 (36): 7–15
Nystrom L, Andersson I, Bjurstam N, Frisell J, Nordenskjold B, Rutqvist LE (2002) Long-term effects of mammography screening: updated overview of the Swedish randomised trials. Lancet 359: 909–919
Nystrom L, Rutqvist LE, Wall S, Lindgren A, Lindqvist M, Ryden S, Andersson I, Bjurstam N, Fagerberg G, Frisell J, Tabar L, Larsson LG (1993) Breast cancer screening with mammography: overview of Swedish randomised trials. Lancet 341: 973–978
Otto SJ, Fracheboud J, Looman CW, Broeders MJ, Boer R, Hendriks JH, Verbeek AL, de Koning HJ, National Evaluation Team for Breast Cancer Screening (2003) Initiation of population-based mammography screening in Dutch Municipalities and effect on breast-cancer mortality: a systematic review. Lancet 361: 1411–1417
Smith RA, Cokkinides V, Brooks D, Saslow D, Brawley OW (2010) Cancer screening in the United States, 2010: a review of current American Cancer Society guidelines and issues in cancer screening. CA Cancer J Clin 60: 99–119
Tabar L, Fagerberg G, Duffy SW, Day NE, Gad A, Grontoft O (1992) Update of the Swedish two-county program of mammographic screening for breast cancer. Radiol Clin North Am 30: 187–210
Tabar L, Yen MF, Vitak B, Chen HH, Smith RA, Duffy SW (2003) Mammography service screening and mortality in breast cancer patients: 20-year follow-up before and after introduction of screening. Lancet 361: 1405–1410
Tan SY, van Oortmarssen GJ, de Koning HJ, Boer R, Habbema JD (2006) The MISCAN-Fadia continuous tumor growth model for breast cancer. J Natl Cancer Inst Monogr 2006 (36): 56–65
US Preventive Services Task Force (2009) Screening for breast cancer: US Preventive Services Task Force recommendation statement. Ann Intern Med 151: 716–726, W-236
Wai ES, D’Yachkova Y, Olivotto IA, Tyldesley S, Phillips N, Warren LJ, Coldman AJ (2005) Comparison of 1- and 2-year screening intervals for women undergoing screening mammography. Br J Cancer 92: 961–966
Warren RM, Young JR, McLean L, Lyons K, Wilson AR, Evans A, Duffy SW, Warsi IM (2003) Radiology review of the UKCCCR Breast Screening Frequency Trial: potential improvements in sensitivity and lead time of radiological signs. Clin Radiol 58: 128–132
White E, Miglioretti DL, Yankaskas BC, Geller BM, Rosenberg RD, Kerlikowske K, Saba L, Vacek PM, Carney PA, Buist DS, Oestreicher N, Barlow W, Ballard-Barbash R, Taplin SH (2004) Biennial vs annual mammography and the risk of late-stage breast cancer. J Natl Cancer Inst 96: 1832–1839
Acknowledgements
This study was supported, in part, by grants 1U01CA152958 and 2U01CA088283 from the National Cancer Institute and by the National Institute for Public Health and the Environment (RIVM).
Author information
Authors and Affiliations
Corresponding author
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
APPENDIX
APPENDIX
This appendix consists of three parts:
-
1)
a model overview containing a description of MISCAN-Fadia,
-
2)
a description of the model components, and
-
3)
a description of the model quantifications of each model component
Model overview
MISCAN-Fadia (MIcrosimulation of SCreening ANalysis – Fatal diameter) is a microsimulation model generating independent life histories. It uses the so-called parallel universe approach and first simulates the individual life histories for women in the absence of screening and then assesses how these histories would change as a consequence of a screening program. A certain percentage of the modelled population develops pre-clinical disease. The natural history of breast cancer is modelled as a continuously growing tumour. MISCAN-Fadia includes a sub model for ductal carcinoma in situ (DCIS), with three different types of preclinical DCIS: regressive DCIS, DCIS that will be diagnosed clinically, and DCIS that will progress to invasive disease. When a screening program is applied, the pre-clinical tumour may be detected by screening if it is larger than a screen-detection threshold diameter.
Model components
Demographics
The demography part of the model simulates individual life histories without breast cancer to form a population. For each person, a date of birth and a date of death of other causes than breast cancer are simulated. The distribution of births and deaths can be adjusted to represent the population simulated.
Incidence
A certain percentage of the modelled population develops preclinical disease. This percentage varies between birth cohorts, while the cohorts have the same age distribution of onset of breast cancer.
Natural history
Among women who develop disease, the natural history of breast cancer is modelled as a continuously growing tumour. Each tumour has a size (the fatal diameter, which differs between tumours) at which diagnosis and treatment will no longer result in cure given available treatment options. If the tumour is diagnosed (either on the basis of clinical presentation with symptoms or by screening) and treated before the tumour reaches the fatal diameter, the woman will be cured and will die of non-breast cancer causes (Appendix Figure A1). Variation between tumours is modelled by probability distributions of tumour growth rate, threshold diameter of screen detection, clinical diagnosis diameter, fatal disease diameter, and survival duration since fatal diameter.

The MISCAN-Fadia natural history model. The model is illustrated by a woman who is diagnosed with incurable breast cancer and for whom screening could have been beneficial. The natural history of breast cancer is simulated through the random selection of six variables from probability distributions, denoted by the various curves: onset=age at tumour onset, growth rate=tumour growth rate, survival=duration between the moment at which the tumour reaches the fatal diameter and the moment of death from breast cancer (not shown), clinical diagn diam=tumour diameter at which the tumour will be diagnosed clinically because of the primary tumour, fatal diam=tumour diameter at which available treatment options will no longer result in cure, threshold diam=tumour diameter at which the tumour becomes screen detectable. After onset the tumour starts growing exponentially according to the tumour growth rate. The diagnosis results from the clinical diagnosis diameter combined with the tumour growth rate. If the tumour is diagnosed after it has reached the fatal diameter, the woman will die from breast cancer. Survival is modelled since fatal diameter. For observed survival (shown), the time between clinical diagnosis and the moment the tumour has reached its fatal diameter has to be subtracted. Screening can change this natural history: After the tumour has reached the threshold diameter, the tumour can be screen detected. If the tumour has not reached the fatal diameter yet at the moment of screen detection, the woman will be cured. Otherwise, screening will not affect the woman's age of death. Reprinted from Tan et al, 2006 with permission from Oxford University Press.
Screening
When a screening program is applied, the preclinical tumour may be detected by screening. Each simulated tumour has a diameter at which it will be clinically diagnosed and a screen-detection threshold diameter. For the latter, screening test sensitivity is 0% below and 100% above this diameter. The threshold diameter is dependent on the calendar year and age of the woman (decreasing with calendar year and older age). Screening benefits result from detection of more tumours at a non-fatal size. The characteristics of organized screening programs, such as screening ages, screening interval and attendance can be specified, and the type of screening (e.g., ‘organized’ or ‘opportunistic’) can be defined in the model.
Treatment
The benefit of adjuvant treatment is modelled as a shift in the fatal diameter for treated women. For each adjuvant treatment a cure proportion is estimated (depending on age). These cure proportions are then translated into corresponding fatal diameters (i.e., a more effective treatment can cure a larger tumour). The dissemination of adjuvant treatment is modelled as the probability of being treated with a certain type of treatment (e.g., chemotherapy, tamoxifen).
Model quantification
Demographics
A female population born between 1930 and 1942 (thus, age 50–62 in 1992) was simulated. We assumed that all cohorts were represented in equal proportions. Each woman is assigned a date of death due to non-breast cancer causes based on the UK female population cohort life tables. These life tables were available from the Human Mortality Database (Human Mortality Database, www.mortality.org). The simulated woman dies because of breast cancer or of other causes, whichever comes first.
Incidence
We used age-specific breast cancer incidence rates for the years 1975–1988, i.e. before the implementation of the National Health Service Breast Screening Programme to estimate age-specific onset parameters for the onset of breast cancer.
Natural history
All parameters were previously estimated using detailed data from the Two County Study (Tabar et al, 1992; Tan et al, 2006). Subsequently, the fatal diameter was calibrated to U.S. data concerning 1975 stage distribution and 1975 survival (SEER data) (Tan et al, 2006).
For the present study, we re-estimated two parameters: the diameter at clinical diagnosis and the screening threshold diameters. We re-estimated these parameters, because these parameters can vary across countries and over time, whereas other parameters (e.g., the tumour growth rate) are assumed to be more or less universal. To estimate these two parameters we used the following data from the UK Frequency Trial:
-
The number of detected invasive cancers over time in both groups (control & study group).
-
The total number of invasive cancers by tumour size and detection mode in both groups.
The estimated values of the diameter at clinical diagnosis and the screening threshold diameters are shown in Appendix Table A1.
The estimated values for these parameters are somewhat higher than the values previously found based on data from the Two County Study (Lognormal (0.8, 0.6) and Weibull (1.0, 3.0) for the diameter at clinical diagnosis and the screening threshold diameter, respectively) (Tan et al, 2006). If these lower values are used to simulate the Frequency Trial, a more favourable tumour size is simulated than the one that is observed in the trial.
Screening
In the present study we simulated the screening in the UK Breast Screening Frequency Trial using previously published attendance rates. The attendance rate in the control group, among women who had attended the prevalence screen, was 85%. In the study group, attendance rates at the three yearly screens were 78, 78, and 81%, respectively (Breast Screening Frequency Trial Group, 2002).
Treatment
We used treatment effectiveness data based on meta-analyses of the Early Breast Cancer Trialists’ Collaborative Group (Early Breast Cancer Trialists’ Collaborative Group, 1998a, 1998b). The dissemination of adjuvant treatment (the probability of being treated with a certain type of treatment) was based on data from the US (Mariotto et al, 2006).
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
van Ravesteyn, N., Heijnsdijk, E., Draisma, G. et al. Prediction of higher mortality reduction for the UK Breast Screening Frequency Trial: a model-based approach on screening intervals. Br J Cancer 105, 1082–1088 (2011). https://doi.org/10.1038/bjc.2011.300
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2011.300
Keywords
This article is cited by
-
Implementation and process evaluation of three interventions to promote screening mammograms delivered for 4 years in a large primary care population
Translational Behavioral Medicine (2017)
-
Is mammography screening history a predictor of future breast cancer risk?
European Journal of Epidemiology (2015)
-
Estimating breast cancer mortality reduction and overdiagnosis due to screening for different strategies in the United Kingdom
British Journal of Cancer (2014)
