
Abstract
The development of new routes for the production of thermoelectric materials with low-cost and high-performance characteristics has been one of the long-term strategies for saving and harvesting thermal energy. Herein, we report a new approach for improving thermoelectric properties by employing the intrinsically low thermal conductivity of a quasi-one-dimensional (quasi-1D) crystal structure and optimizing the power factor with aliovalent ion doping. As an example, we demonstrated that SbCrSe3, in which two parallel chains of CrSe6 octahedra are linked by antimony atoms, possesses a quasi-1D property that resulted in an ultra-low thermal conductivity of 0.56 W m−1 K−1 at 900 K. After maximizing the power factor by Pb doping, the peak ZT value of the optimized Pb-doped sample reached 0.46 at 900 K, which is an enhancement of 24 times that of the parent SbCrSe3 structure. The mechanisms that lead to low thermal conductivity derive from anharmonic phonons with the presence of the lone-pair electrons of Sb atoms and weak bonds between the CrSe6 double chains. These results shed new light on the design of new and high-performance thermoelectric materials.
Similar content being viewed by others

Introduction
The design and engineering of novel thermoelectric materials has been one of the most urgent demands for thermal energy saving and harvesting. The thermoelectric performance is evaluated by the dimensionless thermoelectric figure of merit, ZT=S2σT/(κL+κe+κB), where S, σ, T, κL, κe and κB are the Seebeck coefficient, electrical conductivity, absolute temperature, lattice thermal conductivity, electronic thermal conductivity and bipolar thermal conductivity, respectively. As implied by the definition, a high ZT material must have a large power factor (S2σ) in conjunction with a low thermal conductivity. Unfortunately, except for κL, the other transport parameters (S, σ and κe) are all correlated with the carrier concentration and improving one of them usually leads to diminishing the others. Apparently, a strategy that is based on the reduction of κL is direct and has often been found to be effective. Major reductions in the lattice thermal conductivity have been achieved via point-defect scattering,1, 2 which employs all-scale hierarchical architectures3 and nanostructure engineering.4, 5 Recently, many new materials with high ZT values, owing primarily to their intrinsically low κL, have been found, including Cu3SbSe3,6 Cu2Se,7 SnSe,8 CdSb,9 InTe10 and Cu12Sb4S13.11
Exploring the origin of the intrinsically low lattice thermal conductivity and linking it to the nature of chemical bonding and peculiarities in the crystal structure are not only of fundamental interest but also have major implications regarding the practical aspects of the thermoelectric energy conversion.12 Previous investigations, which have searched for low thermal conductivity compounds, focused on complex crystal structures and materials that have a large unit-cell volume and a large molecular weight.13 However, the chemical bond and atomic coordination environment have not been significantly stressed. In practice, the chemical bonding strength, which is reflected by the Debye temperature, determines the average group velocity of acoustic phonons and the bulk modulus of the compounds.14 Weak chemical bonding generally results in the slow transport of phonons and thus yields lower values of the lattice thermal conductivity.15 Moreover, the asymmetrical local atomic coordination with a large distortion structure, which possess both strong and weak bond types, is beneficial for increasing the bond anharmonicity, which is another important aspect that affects the lattice thermal conductivity.16 The bond anharmonicity serves as a measure of the deviation in lattice vibrations from a perfect harmonic motion and is characterized by the effective Grüneisen parameter. Generally, structures with a chemical bond hierarchy17, 18, 19, 20, 21 and anisotropic multicenter bonding features9 or those that possess a lone pair of electrons22, 23, 24, 25, 26 exhibit a stronger anharmonicity. Therefore, the investigation of the chemical bonding, crystal structures and corresponding lattice dynamics is helpful in understanding phonon transport and the design of materials with an intrinsically low lattice thermal conductivity and high thermoelectric performance.
Low-dimensional materials with a low-dimensional crystal structure, which naturally possess mixed bond types and possibly a local asymmetrical bond environment, offer a broad platform on which to explore new thermoelectric materials with intrinsically low thermal conductivities. Recently, new high-performance thermoelectric materials with anisotropic-layered crystal structures, such as BiCuSeO,27 SnSe,28, 29 Bi2Te330 and Cr2Ge2Te6,31 were shown to possess low lattice thermal conductivities that resulted from their weak chemical bonds and bond anharmonicity. Earlier in 1992, Hicks and Dresselhaus32 pointed out that a reduced dimensionality might have a vital role in reducing the thermal conductivity. This hypothesis has been confirmed by a report on Si nanowires that claimed a high ZT of ~0.6 at 300 K.33 Since then, low dimensionality has been envisioned as a new direction for designing thermoelectric materials with high-performance and much effort has been expended on fabricating low-dimensional materials. Setting aside one-dimensional (1D) nanowires, which have limited applications as power-generating thermoelectric devices, among the best quasi-1D thermoelectric structures are In4Se3 single crystals. Here, the Peierls distortion contributes to a high ZT of 1.4 that was measured along the b axis because of the high Seebeck coefficient, low thermal conductivity and well-controlled electrical conductivity.34 Given the aforementioned encouraging results, searching for novel 1D compounds or optimizing the thermoelectric performance of the existing 1D compounds is worthy of further investigation.
Regarding these issues, we propose an engineering process using the crystallographic structural design and selection of 1D characteristics, bonding anharmonicity by weak-strong bonding architectures and special electronic structures. These crystallographic characteristics and electronic structures are potential candidates with intrinsic low thermal conductivity. By further optimizing the electrical properties through aliovalent ion doping, it is expected that high-performance thermoelectric materials can be attained.
Following these strategies, for example, the crystalline structure of SbCrSe3 can be described and selected as a 1D structural candidate with CrSe6 double chains that are aligned parallel to the b axis.35 Long-range ferromagnetic ordering was observed at temperatures below Tc=72 K. The quasi-1D structure and the semiconducting nature of transport compelled us to explore the thermoelectric properties of this compound. In this work, for the first time, we report on the synthesis, high-temperature thermoelectric properties and the calculated electronic structure of the quasi-1D SbCrSe3 compound, which also shows an intrinsically low thermal conductivity (κL≈1Wm−1 K−1 at 300 K). Using first-principle calculations, we concluded that the presence of strongly anharmonic phonons mainly originated from the presence of the lone electron pairs of Sb atoms. The resulting low thermal conductivity benefitted from the large anharmonicity and weak chemical bonds between the neighboring chains, which yielded low phonon velocities. By intentionally substituting trivalent Sb with divalent Pb, the power factor was greatly enhanced, which led to a peak ZT=0.46 at 900 K in the nominal Pb0.05Sb0.95CrSe3 sample. This value is ~24 times larger than the ZT value that was measured in pristine SbCrSe3. The promising ZT value in doped SbCrSe3 highlights the role of and need for novel 1D compounds for thermoelectric applications.
Experimental procedures
Sample preparation
Polycrystalline PbxSb1-xCrSe3 (x=0, 0.01, 0.03 and 0.05) compounds were synthesized using conventional solid-state sintering. Stoichiometric amounts of high-purity (5N) Sb, Cr, Pb and Se were weighed in a glove box, sealed in a quartz ampoule under a vacuum of 10−6 mbar and then slowly heated to 973 K. The ampoule was maintained at that temperature for 30 h and then furnace-cooled to room temperature. The process was repeated several times to obtain a single phase. The ingot was ground into a powder and sintered using spark plasma sintering (SPS) at 873 K for 10 min under 50 MPa. The relative density of the SPS-sintered pellets was over 98% as measured by the Archimedes’ method. The cylindrical ingots were cut into wafers of 10 mm in diameter and ~1.5 mm in thickness for the thermal conductivity measurements. The thermal diffusivity λ was measured using a Netzsch laser flash diffusivity instrument (LFA-457) under flowing argon. To maximize the laser light absorption, the samples were spray-coated with a thin layer of graphite before the measurements. The thermal conductivity κ was calculated by the equation κ=λ × Cp × d, where Cp and d are the specific heat and mass density, respectively. The specific heat was acquired from a Netzsch DSC404 apparatus (Supplementary Figure S1). Samples of 2.5 × 2.5 × 9.0 mm3 were cut for the simultaneous measurements of the Seebeck coefficient and electrical conductivity using a Linseis instrument (LSR-3) under an atmosphere of helium from room temperature to 920 K. Measurements of the thermal and electronic transport properties were carried out in the same crystallographic direction. The estimated systematic errors in our electrical and thermal transport measurements were 5 and 10%, respectively. The phase, morphology and crystal structure were characterized using powder X-ray diffraction analysis, scanning electron microscopy and transmission electron microscopy. Powder X-ray diffraction analysis was conducted using a PANalytical X-ray diffractometer that was operated at 45 kV and 40 mA. The Rietveld refinements were performed using the TOPAS Academic V4.1-beta (Bruker AXS: Karlsruhe, Germany) software package. The morphology and microstructure were investigated using field emission scanning electron microscopy (JSM-7800F, JEOL) and transmission electron microscopy (JEOL 2100F). The band-gap was obtained by an optical absorption method using a computer-controlled Shimadzu UV-3100 PC that was equipped with an integrating sphere. The sample was ground into a fine powder and compacted on a flat surface of BaSO4 that was used as a 100% reflectance reference material. The bandgap was determined from the tangent line of the absorption edge in the F(R) vs hν plot. Ultrasonic measurements were performed at room temperature to obtain the longitudinal and transverse sound velocities. The source input was generated by a Panametrics 5052 pulser/receiver with the filter set at 0.03 MHz. The response was recorded via a Tektronic TDS5054B-NV digital oscilloscope.
Computational details
The initial crystal structure of SbCrSe3 was optimized using the Vienna ab initio simulation package that was based on density functional theory (DFT).36, 37 Considering the ferromagnetic exchange interaction in SbCrSe3, a ferromagnetic model was built for all calculations. A 1 × 4 × 1 supercell was set up to model calculations of Pb-doped SbCrSe3 with a dopant concentration of 6.25%. The Perdew–Burke–Ernzerhof generalized gradient approximation38 and projector augmented-wave potentials of Blöchl in the implementation of Kresse and Joubert were used.39, 40 Plane waves were included up to the kinetic energy cutoff of 300 eV. For the Brillouin zone (BZ) integration, a 9 × 9 × 9 Monkhorst Pack special k-point grid was applied.41 The stopping criterion for electronic self-consistent interactions was the convergence of the total energy within 10−6 eV. The calculations were performed by allowing both the atomic positions and lattice parameters to find the most stable lattice configuration. The lattice structure was considered to be at equilibrium when the Hellmann−Feynman force on each ion was less than 0.001 eV Å−1. The band energies Ek were computed on a discrete k mesh along the high-symmetry directions, that is, from the BZ center with coordinates Γ (0,0,0) to the F point (0.0,0.5,0.0), Q point (0.0,0.5,0.5), Γ point (0,0,0), Z point (0.0,0.0,0.5), A point (0.5,0.0,0.5), Γ point (0.0,0.0,0.0), B point (0.5,0.0,0.0), C point (0.5, 0.5,0.0) and Γ point (0,0,0) in units of 2π/a, 2π/b and 2π/c. The lattice dynamics of SbCrSe3 was investigated using the frozen-phonon method, which was implemented in the Phonopy package.42 The interactions between the ions and electrons were described by the all-electron projector augmented-wave method with plane waves up to a cutoff energy of 600 eV. The atomic configurations 5s25p3 for Sb, 3d44s2 for Cr and 4s24p4 for Se atoms were treated as the valence electrons. Supercells with dimensions of 2 × 3 × 1 (18.76 Å × 11.53 Å × 13.46 Å, 120 atoms) were used for SbCrSe3. High-symmetry points in the BZ (X(1/2,0,0), Γ(0,0,0), Y(0,1/2,0), P(1/2,1/2,0), Γ(0,0,0) and A(1/2,0,1/2)) were considered in the phonon dispersion and Grüneisen parameter calculations. Atomic displacement parameters were calculated based on the partial phonon density of states from DFT calculations. The Grüneisen parameter characterizes the relationship between the phonon frequency and volume change and thus provides an estimate of the anharmonicity strength. To obtain the mode Grüneisen parameter, we calculated the phonon dispersion in an equilibrium unit-cell volume V and an isotropically expanded volume 1.03 V. The Grüneisen parameter was estimated using the formula:  . The lattice thermal conductivity was calculated based on the Debye–Callaway formalism.43, 44, 45
. The lattice thermal conductivity was calculated based on the Debye–Callaway formalism.43, 44, 45
Results and discussion
Crystal structure
Figures 1a and b show the SbCrSe3 structure that crystallized in the orthorhombic lattice with the lattice parameters a=9.379 Å, b=3.844 Å and c=13.467 Å. The crystal lattice can be viewed as a quasi-1D structure with double chains of CrSe6 octahedra that were aligned parallel to the b axis. Figure 1c shows that the Cr–Se bond distance in these octahedra ranged from 2.5 to 2.6 Å, which is comparable to the distances that were reported for other Cr–Se-based compounds, such as AgCrSe2.46 Within the double chain, the Cr cations formed an edge-sharing triangular arrangement, which led to potential magnetic frustration. The Sb atoms linked the adjacent chains, which resulted in the pseudo-1D crystal structure. There are two types of Sb–Se bonds that are based on the bond distance. In Figure 1d, in the first type, the Sb atom occupies a trigonal pyramidal position relative to the three Se atoms on one end of the double chain. Here, the bond length is ~2.6–2.7 Å, and such a distance is short enough to support the crystal structure. The second bond between Sb and Se is longer at 3.218 Å and couples the Sb and Se atoms on the adjacent chains. Considering that the oxidation state of Sb in SbCrSe3 is 3+, the Sb atoms share their 5p orbitals with Se atoms and the two 5s electrons can be considered as a non-bonding lone electron pair giving rise to anharmonicity and a large Grüneisen parameter. Consequently, SbCrSe3 is expected to exhibit an intrinsically low lattice thermal conductivity.

(a) A perspective view of the crystal structure of SbCrSe3 along the b axis. Brown, blue and green balls indicate Sb, Cr and Se atoms, respectively. (b) Sub-nanotubes (CrSe6 double chains) along the b axis. (c, d) The coordination status of Cr and Sb atoms, respectively.
Phase characterization
For thermoelectric transport characterization, it is crucial to ensure that the quasi-1D structure of SbCrSe3 is a single-phase material because even a small amount of impurity can significantly influence the electronic and thermal transport properties. Figures 2a and c display the powder X-ray diffraction analysis patterns of both the pristine and Pb-doped SbCrSe3 compounds. On the basis of the powder X-ray diffraction patterns, all of the SbCrSe3 reflections were indexed to the orthorhombic structure of the space group Pnma (No. 62) with the lattice parameters a=9.142(1) Å, b=3.785(5) Å and c=13.429(2) Å. No secondary phases were detected. The Rietveld refinement was conducted to obtain the actual lattice parameters. Our data are in good agreement with previously reported data. The lattice parameters of Pb-doped samples are shown in Figure 2b. Compared with the values for pure SbCrSe3, the lattice parameter a slightly decreased, whereas the parameters b and c increased, which results in an overall slightly increased unit-cell volume V. This increase is likely due to the weakened Sb–Se bond between the double CrSe6 chains. Because the covalent radius of Pb is larger than that of Sb, the lattice parameters b and c increased with increasing Pb content up to x=0.05, which suggests that this is the solubility limit of Pb in SbCrSe3.

(a) XRD patterns of PbxSb1−xCrSe3 (x=0, 0.01, 0.03 and 0.05) samples. (b) Lattice parameters and cell volume of PbxSb1−xCrSe3 (x=0, 0.01, 0.03 and 0.05). (c) XRD patterns of SbCrSe3 samples via Rietveld refinement. XRD, X-ray diffraction analysis.
Electrical transport properties
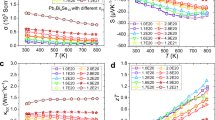
Figure 3 displays the temperature dependence of the electrical transport properties for all PbxSb1-xCrSe3 (x=0.0, 0.01, 0.03 and 0.05) compounds. In Figure 3a, SbCrSe3 exhibits a low electrical conductivity at room temperature but increases with increasing temperature because of the thermal activation of the intrinsic carriers. In the Pb-doped SbCrSe3 compounds, Pb behaved like an effective p-type dopant and each Pb atom that substituted for Sb was expected to contribute one hole to the valence band of SbCrSe3. Consequently, Pb doping led to a markedly enhanced electrical conductivity. The electrical conductivity σ significantly increased from 0.054 S m−1 at 300 K in the pristine SbCrSe3 to 1586.1 S m−1 at 300 K for the Pb-doped sample at x=0.05. Figure 3b shows the Hall carrier concentration p and carrier mobility μ for the Pb-doped compounds at room temperature. Pristine SbCrSe3 has a low hole carrier concentration of p~2.2 × 1016 cm−3. As expected, the concentration of holes dramatically increased upon doping with Pb and reached p~0.9 × 1019 cm−3 and p~4.5 × 1019 cm−3 for compounds with Pb contents of x=0.01, 0.03 and 0.05, respectively. Although the mobility of holes is low in the range of 1.5–2.3 cm2 V−1 s−1, an interesting rising trend with the content of Pb up to x=0.03 was demonstrated. This trend is likely due to the disruption of localized Cr 3d orbitals upon Pb doping on the Sb site. The mobility values are comparable to those measured on doped MnSb2Se447 but are lower than the mobilities of the state-of-the-art thermoelectric materials BiCuSeO48 and alpha-MgAgSb.49 In SbCrSe3, we ascribed low mobilities to the strong scattering of the charge carriers (holes), the natural low dimensionality of the crystal structure and the spatially localized nature of Cr 3d orbitals.

Transport properties of PbxSb1−xCrSe3 (x=0, 0.01, 0.03 and 0.05) samples. (a) Temperature-dependent electrical conductivity (σ). (b) Hall carrier concentration (black symbols) and carrier mobility (blue symbols) at room temperature. (c) Temperature-dependent Seebeck coefficient (S). (d) Temperature-dependent power factor (S2σ).
The temperature dependence of the Seebeck coefficient is depicted in Figure 3c. This dependence revealed a typical intrinsic semiconductor with an initially increasing and subsequently decreasing magnitude of the Seebeck coefficient because the minority carriers are activated at elevated temperatures. Pb doping has a profound effect on the Seebeck coefficient of SbCrSe3. With a sharply increasing concentration of holes upon Pb doping, the Seebeck coefficients are suppressed compared with SbCrSe3 and the peak is shifted to higher temperatures (or is not observed in the covered range of temperatures). Nevertheless, the Seebeck coefficients remained substantial and, even for a highly doped sample with x=0.05, the magnitude of the Seebeck coefficient at 300 K reached over 160 μV K−1. Similar to the electrical conductivity, the Seebeck coefficient showed an increasing trend with temperature, which suggests that the doped SbCrSe3 samples show a weakly degenerate semiconducting behavior. Benefiting from the increased electrical conductivity and substantial values of the Seebeck coefficient, Pb-doped SbCrSe3 compounds have higher power factors than the pristine SbCrSe3 sample. In Figure 3d, the maximum power factor of 0.30 mW m−1 K−2 was achieved at 900 K for Pb0.05Sb0.95CrSe3, the value being ~22 times larger than the power factor of pristine SbCrSe3. In spite of these impressive enhancements upon Pb doping, the power factor was still low compared to classic thermoelectric materials due primarily to the low carrier mobility and low electrical conductivity.
To better understand the differences in the electrical conductivity between the pristine SbCrSe3 and Pb-doped samples, we analyzed the bandgap energy, density of states (DOSs) effective mass, which was estimated using the single parabolic band model and assumed the dominance of acoustic-phonon scattering, and band structure that was calculated via the Vienna ab initio simulation package tool.
For semiconducting materials, the bandgap energy can be estimated by the Goldsmid–Sharp formula50 Eg=2eSmaxTmax, where Smax is the maximum Seebeck value and Tmax is the temperature at which this maximum occurs. This formula yields Eg=0.57 eV for undoped SbCrSe3. This value is slightly smaller than the UV-optics value of 0.7 eV (Supplementary Figure S2) and larger in comparison with the calculated DFT value of 0.4 eV. It is common to see significantly smaller values of the bandgap based on DFT calculations owing to deficiencies in the Perdew–Burke–Ernzerhof exchange correlation function. To obtain theDOSs effective mass m* and present a quantitative understanding of the microscopic mechanism that governs the Seebeck coefficient in Pb-doped samples, the single parabolic band model was utilized. For the Pb-doped samples, an estimate of the Seebeck coefficient can be obtained using the following equations:

In equation (1), n is the nth order of the Fermi integral, η is the reduced Fermi energy, r is the scattering factor, h is the Planck’s constant, kB is the Boltzmann constant and e is the electron charge. Note that in this model, m* is equivalent to theDOSs effective mass usually designated as m*DOS. Considering that the dominant scattering mechanism for most thermoelectric materials at elevated temperatures is acoustic-phonon scattering, r was taken as equal to −1/2 in all data fittings. The carrier concentration dependence of the Seebeck coefficient is illustrated by the Pisarenko plot in Figure 4. The solid curves were generated at 300 K with an effective mass of m*=0.61 me for the sample with a Pb content of x=0.01 and an effective mass m*=1.0 me for samples with a Pb content of x=0.03 and x=0.05. In principle, the increased m* may be due to the convergence of closely lying valence band edges, which demonstrates that the single parabolic band model may not be used to depict the electrical transport properties. In addition, band structure calculations of SbCrSe3 present a multi-valley dispersion at the top of the valence band.

The Pisarenko plots (Seebeck coefficient vs Hall carrier density) at 300 K with a fixed DOSs effective mass m*DOS.
To gain further insight into the electronic behavior, we calculated the band structure of SbCrSe3 and a Pb-doped sample with x=0.065. Since the transport properties are mostly sensitive to the energy states near the Fermi level, we have focused solely on the electronic states near the valence band maximum. In Figures 5a and b, the band structures of SbCrSe3 and Pb0.0625Sb0.9375CrSe3 featured an indirect bandgap with the valence band maximum in the middle of the Γ-Z symmetry direction and the CBM (conduction band minimum) at Z. In Pb0.0625Sb0.9375CrSe3, the Fermi level clearly shifted deeper into the VB and the energy gap between the first and second valence bands slightly increased. As a result, the electrical conductivity was greatly enhanced by Pb doping owing to the large increase in carrier concentration. Figures 5c and d display the DOSs of SbCrSe3 and Pb0.0625Sb0.9375CrSe3. In the pristine SbCrSe3 compound, the Sb atoms slightly contributed to the valence band maximum near the Fermi level. The primary role of Sb is to donate electrons, saturate the chemical bond and stabilize the compound. An important feature that determines the band-edge property was strong hybridization of the Cr-d and Se-p orbitals in the upper VB states. This d–p orbital hybridization was also manifested by the partial charge densities (Supplementary Figure S3a). In the Pb0.0625Sb0.9375CrSe3 compound, the Pb-p and Se-p anti-bonding states dominated at the top of the valence band and the hybridized Cr-d and Se-p orbitals became weakly delocalized. The Pb-p and Se-p anti-bonding states originated from both within-the-chain and inter-chain interactions (Supplementary Figure S3b) and strengthened the carrier conducting network. These active anti-bonding states linked the individual chains within the double chains and led to a locally three-dimensional crystalline environment with an increase in electrical conductivity.

(a) The band structure of SbCrSe3. (b) The band structure of Pb0.0625Sb0.9375CrSe3. (c) The total and partial density of states of SbCrSe3. (d) The total and partial density of states of Pb0.0625Sb0.9375CrSe3.
Thermal transport properties
Figure 6a shows the total thermal conductivity of PbxSb1−xCrSe3 as a function of temperature. The total thermal conductivity κ can be divided into contributions from the lattice κL and charge carrier κe, which are referred to as the electrical thermal conductivity. Assuming a constant Lorenz number L=2.45 × 10−8 V2 K−2, κe can be estimated by the Wiedemann–Franz law (κe=LσT). Consequently, κL can be readily obtained by subtracting κe from κ. In both doped and undoped PbxSb1−xCrSe3, κe is small (<0.08 W m−1 K−1) (Figure 6b) because of the relatively low electrical conductivity. In Figure 6c, owing to Umklapp scattering, κL decreased with increasing temperature up to ~800 K, at which point the bipolar contribution was noticeable. The thermal diffusivity λ showed a negative temperature dependence below 800 K, whereas above this temperature intrinsic excitations dominated. The SbCrSe3 compound exhibited low lattice thermal conductivities of 1.08 W m−1 K−1 at 300 K and 0.56 W m−1 K−1 at 900 K, which are lower than those of the other PbTe-based materials.51 Figure 6d displays the ZT values of PbxSb1−xCrSe3. The peak ZT of undoped SbCrSe3 was 0.018 at 800 K. The doped samples exhibited much higher ZTs and the peak value was shifted towards higher temperatures. The largest ZT=0.46 was achieved at 900 K in Pb0.05Sb0.95CrSe3. This value is 24 times larger than the figure of merit of pristine SbCrSe3. Such increased ZT values in the doped compounds were attributed to cooperatively acting large enhancements in the electrical conductivity and the decreased lattice thermal conductivity upon Pb doping.

(a) Total thermal conductivity; (b) the lattice thermal conductivity; (c) the electronic thermal conductivity; (d) ZT values for SbCrSe3 and PbxSb1-xCrSe3 (x=0.01, 0.03 and 0.05).
Considering that the room-temperature lattice thermal conductivity of SbCrSe3 is 1.08 W m−1 K−1 and that of Pb0.05Sb0.95Cr5Se8 is 0.78 W m−1 K−1, Pb doping depressed κL by 28%. This reduction indicates that the Pb/Sb disorder in the Sb sublattice strongly enhanced the phonon scattering (details are in the Supporting Information). In Figure 7a, the experimental data followed the same trend that was predicted by the model, although the experimental data are to some degree higher than the calculated values. Γmass and Γstrain are plotted in Figure 7b. It is clear that the value of Γstrain is much larger than that of Γmass. This demonstrates that strain field fluctuations, which were introduced by substituting Pb in the Sb sublattice, are the dominant factor in the point-defect scattering of phonons and led to a significantly decreased lattice thermal conductivity in Pb-doped SbCrSe3 compounds. The large strain field fluctuation contribution is related to the local structural distortion between the double-ladder chains. Figure 7c shows the local coordination environment of the Sb and Pb atoms between the ferromagnetism chains of SbCrSe3 (left) and Pb0.0625Sb0.9375CrSe3 (right), which was based on the DFT optimization. In SbCrSe3, the Sb atom was coordinated with three Se atoms and formed almost equidistant bond lengths (2.689 Å, 2.689 Å and 2.714 Å) that produced a relatively stable triangular pyramid between the double-ladder chains. However, when an impurity Pb atom was introduced, the Pb atoms were more inclined to form bonds with the Se atoms on the neighboring chain, which resulted in the coordination number of the Pb atom of four (3.139 Å, 2.998 Å, 2.818 Å and 2.818 Å). Moreover, the originally relatively stable triangular pyramid became a relatively unstable square pyramid. Therefore, the location of the Pb atom in Pb0.0625Sb0.9375CrSe3 deviated from the original relatively stable location of the Sb atom in SbCrSe3, which led to additional local distortion. The large differences in the coupling forces of a Se atom with Pb and a Se atom with Sb are expected to produce to a strong strain fluctuation contribution in the phonon scattering. A similarly strong strain fluctuation contribution arising from the presence of impurity atoms was also observed in CuFeS252 and Cu2Zn1−xFexGeSe4.53

(a) Comparison of the experimental lattice thermal conductivity and calculated values using the model of Callaway for PbxSb1−xCrSe3 (x=0, 0.01, 0.03 and 0.05) at 300 K. (b) Calculated strain and mass disorder parameter Γ for PbxSb1−xCrSe3 (x=0, 0.01, 0.03 and 0.05) at 300 K. (c, d) Local atomic coordination environment between double CrSe6 chains in SbCrSe3 and in Pb0.0625Sb0.9375CrSe3 obtained via DFT.
The origin of the low thermal conductivity
To shed light on the origin of the intrinsically low lattice thermal conductivity of SbCrSe3, we calculated the phonon dispersion, projected phononDOSs and Grüneisen parameters of the acoustic phonons. Then, we modeled the low-temperature lattice thermal conductivity using the Debye–Callaway model. Figure 8a shows the calculated phonon curves at different unit-cell volumes (V and 1.03V) along the high-symmetry directions in the first BZ. Three phonon dispersions (TA, TA′ and LA), which represent the major contributions to the thermal conductivity, are highlighted in color. The longitudinal acoustic branch (LA) and transverse acoustic branch (TA′) interacted with the low-lying optical branches along multiple directions at a location approximately midway to the zone boundary. An obvious avoided-crossing is observed because of the lowest frequency of 28 cm−1 in the optic modes. The frequencies of the optic-phonon branch are lower than the vibrational frequency of the La ions in filled skutterudites.54 Such a low optic-mode frequency was also found in the intrinsically low thermal conductivity of the Cu3SbSe3 and CdSb compounds.9, 17 The TA branch exhibited normal behavior with no interactions with the optic-phonon branches, and its frequencies smoothly expanded toward the zone boundary and saturated at ~20–40 cm−1. The acoustic and low-frequency optic-phonon branches, which were responsible for enhanced scattering and low lattice thermal conductivity, originated from the (Sb–Se) group, as indicated in Figure 8b. In the phonon DOS, the low frequency branches were mainly associated with the vibrations of Sb and Se atoms, which indicate that the Sb–Se bond is crucial to heat transport in SbCrSe3. Therefore, the chemical bond strength and the local environment of Sb–Se should be closely studied.

(a) Phonon dispersion in SbCrSe3 along high-symmetry lines. (b) Total phonon density of states and partial contributions from Sb, Cr and Se. (c) Calculated ADPs and the corresponding atom projections for different atoms in SbCrSe3 based on the phonon spectrum. (d) Mode Grüneisen parameters of the acoustic branches along high-symmetry lines for the LA, TA and TA' vibrations. (e) Average partial Grüneisen parameters for the LA, TA and TA' mode vibrations in SbCrSe3. ADP, atomic displacement parameter.
The atomic displacement parameter is defined as the mean square amplitude of the vibrations of an atom around its equilibrium position and reflects the strength of the chemical bond. Generally, a relatively large atomic displacement parameter suggests that the atom in question vibrates with a larger amplitude from its equilibrium position than the other atoms. This means that the restoring force is weak because the chemical bond is weak.18 In Figure 8c, the atomic displacement parameters of the Sb and Se atoms are larger than that of the Cr atom and the values in the X and Z directions are higher than those in the Y direction. In particular, the calculated atomic displacement parameter values for the Sb atom in the Z direction are approximately two times those of the other atoms in SbCrSe3, which documents a chemical bond hierarchy in its neighboring environment. Weak bonds that are distributed along the double CrSe6 chain result in both a low sound velocity and Young's modulus.
The group velocities and Debye temperatures of the three acoustic-phonon branches were calculated and are listed in Table 1. The average sound velocity was estimated to be 2037 m s−1, which is consistent with the experimental result of 2092 m s−1. The calculated results were similar to the value for BiCuSeO (2107 m s−1),48 larger than those of PbTe (1770 m s−1),51 and smaller than that of the Zintl chain compound Ca3AlSb3 (2710 m s−1).55 Materials with a low Young's modulus are considered to have ‘soft’ bonding, which results in slow phonon transport and thus a low lattice thermal conductivity. The calculated Young's modulus48 of SbCrSe3 is 64 GPa, which is similar to those of the low lattice thermal conductivity compounds CdSb (55–60 GPa, 1590 m s−1, 1.0 W m−1 K−1)9 and BiCuSeO (76.5 GPa, 2107 m s−1, 0.9 W m−1 K−1).48 We have also plotted the dispersion of the Grüneisen parameters of the acoustic modes in SbCrSe3. The Grüneisen parameter provides an estimate of the anharmonicity strength in a compound and influences the phonon–phonon Umklapp processes that limit the lattice thermal conductivity. From Figure 8d, we found that different phonon branches possessed different Grüneisen parameters throughout the first BZ. Remarkably, unusually high values of the Grüneisen parameter were observed near the zone boundary and close to the midpoint between the zone center and boundary. This indicates that the three acoustic-phonon branches are anharmonic. To clarify the origin of anharmonic phonon interactions in SbCrSe3 at low temperatures, a partial Grüneisen parameter for each atom type was estimated using the method described by Qiu et al.,17, 18 and the results are shown in Figure 8e. The average Grüneisen parameter of SbCrSe3 is heavily weighted by contributions of the Sb atom, which implies that Sb atoms play a dominant role in intrinsic phonon–phonon interactions at low temperatures and can be traced to the bond chemical hierarchy in the neighborhood of the Sb3+ ion and its lone pair of 5s electrons. On the basis of the above discussion, we attributed the low lattice thermal conductivity of SbCrSe3 to a large anharmonicity of the structure and a relatively low average sound velocity. Given the phonon group velocities, average mode Grüneisen parameters and Debye temperatures for the three acoustic-phonon branches, we utilized the Debye–Callaway model and calculated the lattice thermal conductivity. The experimental and theoretical results agree reasonably well in magnitude, but the experimental data show somewhat weaker temperature dependence (Supplementary Figure S4).
Conclusions
In this study, phase-pure PbxSb1-xCrSe3 (x=0, 0.01, 0.03 and 0.05) compounds have been successfully synthesized, and their high-temperature thermoelectric properties were investigated for the first time. SbCrSe3 exhibited an intrinsically low lattice thermal conductivity that was attributed to the bond anharmonicity, lone-pair electrons of Sb and weak bonding between the CrSe6 double chains, which resulted from the quasi-1D structure. The electronic structure calculations suggest that the valence band edge was dominated by Cr-d and Se-p orbitals. The Pb doping on Sb sites produced a noticeable increase in the electrical conductivity, whereas a relatively large Seebeck coefficient was maintained. A combination of an intrinsically low thermal conductivity and a substantially enhanced power factor led to a ZTmax=0.46 at 900 K in Pb0.05Sb0.95CrSe3. This work demonstrates that bulk compounds with the quasi-1D structure are good candidates for thermoelectric materials and sheds new light on the design of and search for new high-performance thermoelectric materials.
References
Li, Z., Xiao, C., Fan, S. J., Deng, Y., Zhang, W. S., Ye, B. J. & Xie, Y. Dual vacancies: an effective strategy realizing synergistic optimization of thermoelectric property in BiCuSeO. J. Am. Chem. Soc. 137, 6587–6593 (2015).
Li, Z., Xiao, C., Zhu, H. & Xie, Y. Defect chemistry for thermoelectric materials. J. Am. Chem. Soc. 138, 14810–14819 (2016).
Biswas, K., He, J. Q., Blum, I. D., Wu, C. I., Hogan, T. P., Seidman, D. N., Dravid, V. P. & Kanatzidis, M. G. High performance bulk thermoelectric with all scale hierarchical architectures. Nature 489, 414–420 (2012).
Heremans, J. P., Thrush, C. M. & Morelli, D. T. Thermopower ehancement in lead telluride nanostructures. Phys. Rev. B 70, 115334–115339 (2004).
Hsu, K. F., Loo, S., Guo, F., Chen, W., Dyck, J. S., Uher, C., Hogan, T., Polychroniadis, E. K. & Kanatzidis, M. G. Cubic AgPb(m)SbTe(2+m): bulk thermoelectric materials with high figure of merit. Science 303, 818–821 (2004).
Wei, T. R., Wu, C. F., Sun, W., Pan, Y. & Li, J. F. Is Cu3SbSe3 a promising thermoelectric material? RSC Adv. 5, 42848–42854 (2015).
Liu, H. L., Shi, X., Xu, F. F., Zhang, L. L., Zhang, W. Q., Chen, L. D., Li, Q., Uher, C., Day, T. & Snyder, G. J. Copper ion liquid like thermoelectric. Nat. Mater. 11, 422–426 (2012).
Zhao, L. D., Lo, S. H., Zhang, Y. S., Sun, H., Tan, G. J., Uher, C., Wolverton, C., Dravid, V. P. & Kanatzidis, M. G. Ultra thermal conductivity and high thermoelectric figure of merit in SnSe crystals. Nature 508, 373–377 (2014).
Wang, S., Yang, J., Wu, L. H., Wei, P., Yang, J. H., Zhang, W. Q. & Grin, Y. Anisotropic multicenter bonding and high thermoelectric perfromance in electron poor CdSb. Chem. Mater. 27, 1071–1081 (2015).
Jana, M. K., Pal, K., Waghmare, U. V. & Biswas, K. The origin of ultralow thermal conductivity in InTe: lone pair induced anharmonic rattling. Angew Chem. Int. Ed. Engl. 55, 1–6 (2016).
Lu, X., Morelli, D. T., Xia, Y., Zhou, F., Ozolins, V., Chi, H., Zhou, X. Y. & Uher, C. High performance thermoelectricity in earth abundant compounds based on natural mineral tetrahedrites. Adv. Energy Mater. 3, 342–348 (2013).
Zeier, W. G., Zevalkink, A., Gibbs, Z. M., Hautier, G., Kanatzidis, M. G. & Snyder, G. J. Thinking like a chemist: intuition in thermoelectric materials. Angew Chem. Int. Ed. Engl. 55, 2–18 (2016).
Tritt, T. M. & Subramanian, M. A. Thermoelectric materials, phenomena, and applications: a bird's eye view. MRS Bull 31, 188–220 (2006).
Spitzer, D. P. Lattice thermal conducitity of semiconductors: a chemical bond approach. J. Phys. Chem. Solids 31, 19–40 (1970).
Pei, Y. L., He, J. Q., Li, J. F., Li, F., Liu, Q. J., Pan, W., Barreteau, C., Berardan, D., Dragoe, N. & Zhao, L. D. High thermoelectric performance of oxyselenides: intrinsically low thermal conductivity of Ca-doped BiCuSeO. NPG Asia Mater. 5, e47–e56 (2013).
Yang, J., Xi, L. L., Qiu, W. J., Wu, L. H., Shi, X., Chen, L. D., Yang, J. H., Zhang, W. Q., Uher, C. & Singh, D. J. On the tuning of electrical and thermal transport in thermoelectrics: an integrated theory experiment perspective. NPG Comput. Mater. 2, 15015–15032 (2016).
Qiu, W. J., Xi, L. L., Wei, P., Ke, X. Z., Yang, J. H. & Zhang, W. Q. Part Crystalline part liquid state and rattling like thermal damping in materials with chemical bond hierarchy. Proc. Natl Acad. Sci. USA 111, 15031–15035 (2014).
Qiu, W. J., Wu, L. H., Ke, X. Z., Yang, J. H. & Zhang, W. Q. Diverse lattice dynamics in ternary Cu-Sb-Se compounds. Sci. Rep. 5, 13643–13650 (2015).
Chi, H., Kim, H., Thomas, J. C., Su, X. L., Stackhouse, S., Kaviany, M., Ven, A. V. D., Tang, X. F. & Uher, C. Configuring pnicogen rings in skutterudites for low phonon conductivity. Phys. Rev. B. 86, 195209–195216 (2012).
Tadano, T., Gohda, Y. & Tsuneyuki, S. Impact of rattlers on thermal conductivity of a thermoelectric clathrate: a first principles study. Phys. Rev. Lett. 114, 095501–095506 (2015).
Christensen, M., Abrahamsen, A. B., Christensen, N. B., Juranyi, F., Andersen, N. H., Lefmann, K., Andreasson, J., Bahl, C. R. H. & Iversen, B. B. Avoided crossing of rattler modes in thermoelectric materials. Nat. Mater. 7, 811–815 (2008).
Nielsen, M. D., Ozolins, V. & Heremans, J. P. Lone pair electrons minimize lattice thermal conductivity. Energy Environ.Sci. 6, 570–578 (2013).
Morelli, D. T., Jovovic, V. & Heremans, J. P. Instrinsically minimal thermal conductivity in cubic I-V-VI2 semiconductors. Phys. Rev. Lett. 101, 035901–035905 (2008).
Guin, S. N., Chatterjee, A., Negi, D. S., Datta, R. & Biswas, K. High thermoelectric performance in tellurium free p-Type AgSbSe2 . Energy Environ. Sci. 6, 2603–2608 (2013).
Skoug, E. J. & Morelli, D. T. Role of lone pair electrons in producing minimum thermal conductivity in nitrogen group chalcogenide compounds. Phys. Rev. Lett. 107, 235901–235906 (2011).
Lai, W., Wang, Y. X., Morelli, D. T. & Lu, X. From bonding asymmetry to anharmonic rattling in Cu12Sb4S13 Tetrahedrites: when lone pair electrons are not so lonely. Adv. Funct. Mater. 25, 3648–3657 (2015).
Pei, Y. L., Wu, H. J., Wu, D., Zheng, F. S. & He, J. Q. High thermoelectric performance realized in a BiCuSeO system by improving carrier mobility through 3D modulation doping. J. Am. Chem. Soc. 136, 13902–13908 (2014).
Zhao, L. D., Tan, G. J., Hao, S. Q., He, J. Q., Pei, Y. L., Chi, H., Wang, H., Gong, S., Xu, H. B., Dravid, V. P., Uher, C., Snyder, G. J., Wolverton, C. & Kanatzidis, M. G. Ulatrhigh power factor and thermoelectric performance in hole doped single crystal SnSe. Science 351, 141–149 (2016).
Peng, K. L., Lu, X., Zhan, H., Hui, S., Tang, X. D., Wang, G. W., Dai, J. Y., Uher, C., Wang, G. Y. & Zhou, X. Y. Broad temperature plateau for high zTs in heavily doped P type SnSe single crystals. Energy Environ. Sci. 9, 454–460 (2015).
Saleemi, M., Toprak, M. S., Li, S. H., Johnsson, M. & Muhammed, M. Synthesis, processing and thermoelectric porperties of bulk nanostructured bismuth tellruide. J. Mater. Chem. 22, 725–730 (2012).
Yang, D. F., Yao, W., Chen, Q. F., Peng, K. L., Jiang, P. F., Lu, X., Uher, C., Yang, T., Wang, G. Y. & Zhou, X. Y. Cr2Ge2Te6: high thermoelectric performance from layered structure with high symmetry. Chem Mater. 28, 1611–1615 (2016).
Hicks, L. D. & Dresselhaus, M. S. Effect of quantum well structures on the thermoelectric figure of merit. Phys. Rev. B. 47, 12727–12732 (1993).
Hochbaum, A. I., Chen, R., Delagdo, R. D., Liang, W. J., Garnett, E. C., Najarian, M., Majumdar, A. & Yang, P. D. Enhaced thermoelectric performance of rough silicon nanowires. Nature 451, 163–167 (2008).
Rhyee, J. S., Lee, K. H., Lee, S. M., Cho, E., Kim, S. II, Lee, E., Kwon, Y. S., Shim, J. H. & Kotliar, G. Peierls distortion as a route to high thermoelectric performance in In4Se3-x crystals. Nature 459, 965–968 (2009).
Odink, D. A., Carteaux, V., Payen, C. & Ouvrard, G. Synthesis and structure of chromium antimony triselenide(CrSbSe3: a pseudo one dimensional ferromagnet. Chem Mater. 5, 237–240 (1993).
Kresse, G. & Furthmuller, J. Efficiency of abinitio total energy calculations for metals and semiconductors using a plane wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Kresse, G. & Furthmuller, J. Efficient iterative schemes for abinitio total energy calculations using a plane wave basis set phys. Phys. Rev. B 54, 11169–11186 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximationn made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Blöchl, P. E. Proector augmented wave method. Phys. Rev. B 50, 17953–17979 (1994).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented wave method. Phys. Rev. B 59, 1758–1775 (1999).
Monkhorst, H. J. & Pack, J. D. Special points for Brillouin Zone integrations. Phys. Rev. B 13, 5188–5192 (1976).
Togo, A. & Tanaka, I. First principles phonon calculations in materials science. Script. Mater. 108, 1–5 (2015).
Callaway, J. Model for lattice thermal conductivity at low temperatures. Phys. Rev. 113, 1046–1051 (1959).
Morelli, D. T., Heremans, J. P. & Slack, G. A. Estimation of the isotope effect on the lattice conductivity of group IV and group III-V semiconductors. Phys. Rev. B 66, 195304–195313 (2002).
Zhang, Y. S., Skoug, E., Cain, J., Ozolins, V., Morelli, D. & Wolverton, C. First principles description of anomalously low lattice thermal conductivity in thermoelectric Cu-Sb-Se ternary semiconductors. Phys. Rev. B 85, 054306–054312 (2012).
Damay, F., Petit, S., Rols, S., Braendlein, M., Daou, R., Elkaim, E., Fauth, F., Gascoin, F., Martin, C. & Maignan, A. Localised Ag+ vibrations at the origin of ultralow thermal conductivity in layered thermoelectric AgCrSe2 . Sci. Rep. 6, 23415–23422 (2016).
Li, S. M., Zhao, H. Z., Zhang, H., Ren, G. K., Liu, N., Li, D. D., Yang, C. S., Jin, S. F., Shang, D. S., Wang, W. H., Lin, Y. H., Gu, L. & Chen, X. L. Enhancement of the thermoelectric properties of MnSb2Se4 through Cu resonant doping. RSC Adv. 5, 99065–99073 (2015).
Zhao, L. D., He, J. Q., Berardan, D., Lin, Y. H., Li, J. F., Nan, C. W. & Dragoe, N. BiCuSeO oxyselenides: new promising thermoelectric materials. Energy Environ. Sci. 7, 2900–2924 (2014).
Ying, P. J., Liu, X. H., Fu, C. G., Yue, X. Q., Xie, H. H., Zhao, X. B., Zhang, W. Q. & Zhu, T. J. High performance alpha-MgAgSb thermoelectric materials for low temperature power generation. Chem. Mater. 27, 909–913 (2015).
Gibbs, Z. M., Kim, H. S., Wang, H. & Snyder, G. J. Band gap estimation from temperature dependent seebeck meaasurement deviations from the 2eSmaxTmax relation. Appl. Phys. Lett. 106, 022112–022117 (2015).
Pei, Y. Z., Gibbs, Z. M., Balke, B., Zeier, W. G. & Snyder, G. J. Optimum carrier concentration in n Type PbTe thermoelectrics. Adv. Energy Mater. 4, 1400486–1400498 (2014).
Li, Y. L., Zhang, T. S., Qin, Y. T., Day, T., Snyder, G. J., Shi, X. & Chen, L. D. Thermoelectric transport properties of diamond like Cu1-xFe1+xS2 tetrahedral compound. J. Appl. Phys. 116, 203705–203713 (2014).
Heinrich, C. P., Day, T. W., Zeier, W. G., Snyder, G. J. & Tremel, W. Effect of isovalent substitution on the thermoelectric properties of the Cu2ZnGeSe4-xSx series of solid solutions. J. Am. Chem. Soc. 136, 442–448 (2014).
Feldman, J. L., Singh, D. J., Mazin, I. I., Mandrus, D. & Sales, B. C. Lattice dynamics and reduced thermal conductivity of filled skutterudites. Phys. Rev. B. 61, 9209(R)–9212(R) (2000).
Zevalkink, A., Toberer, E. S., Zeier, W. G., Larsen, E. F. & Snyder, G. J. Ca3AlSb3: an inexpensive non-toxic thermoelectric material for waste heat recovery. Energy Environ. Energy Environ. Sci. 4, 510–518 (2011).
Acknowledgements
This work was financially supported in part by the National Natural Science Foundation of China (grant nos. 11674040, 11404044, 51472036, 51672270, 21671028) and the Fundamental Research Funds for the Central Universities (106112016CDJZR308808). This work was conducted at the Chongqing Institute of Green and Intelligent Technology, Chinese Academy of Sciences, and supported by the 100 Talent Program (Grant No. 2013-46) and the Key Research Program for Frontier Sciences, CAS (QYZDB-SSW-SLH016). CU acknowledges support from the CERC-CVC and the joint US-China Research Program that was supported by the US Department of Energy (DE-PI0000012).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the NPG Asia Materials website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Yang, D., Yao, W., Yan, Y. et al. Intrinsically low thermal conductivity from a quasi-one-dimensional crystal structure and enhanced electrical conductivity network via Pb doping in SbCrSe3. NPG Asia Mater 9, e387 (2017). https://doi.org/10.1038/am.2017.77
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/am.2017.77
This article is cited by
-
Identification of vibrational mode symmetry and phonon anharmonicity in SbCrSe3 single crystal using Raman spectroscopy
Science China Materials (2021)
