
ABSTRACT
Type I interferons (IFN) are well studied cytokines with anti-viral and immune-modulating functions. Type I IFNs are produced following viral infections, but until recently, the mechanisms of viral recognition leading to IFN production were largely unknown. Toll like receptors (TLRs) have emerged as key transducers of type I IFN during viral infections by recognizing various viral components. Furthermore, much progress has been made in defining the signaling pathways downstream of TLRs for type I IFN production. TLR7 and TLR9 have become apparent as universally important in inducing type I IFN during infection with most viruses, particularly by plasmacytoid dendritic cells. New intracellular viral pattern recognition receptors leading to type I IFN production have been identified. Many bacteria can also induce the up-regulation of these cytokines. Interestingly, recent studies have found a detrimental effect on host cells if type I IFN is produced during infection with the intracellular gram-positive bacterial pathogen, Listeria monocytogenes. This review will discuss the recent advances made in defining the signaling pathways leading to type I IFN production.
Similar content being viewed by others

Introduction
The type I interferons (IFN) were the first cytokines discovered and named for their potent ability to “interfere” with viral replication 1. In 1957, Isaacs and Lindenmann reported a secreted factor termed “interferon” that could induce a virus-resistant state in chick cells after influenza virus infection 1. The type I IFN family consists of multiple IFNα members, single IFNβ, ε, κ, and ω subtypes, as well as δ and τ subtypes found in pig and ovine respectively 2. These cytokines induce antiviral responses by binding a common receptor, the type I IFNα/β receptor (IFNAR), expressed on a wide variety of cell types 2.
Over the past few years, our knowledge concerning the mechanism of transcriptional regulation of type I IFNs has rapidly expanded. The pathogen ligands, respective host receptors, and signaling pathways that trigger type I IFN induction are becoming elucidated. While there are still many questions regarding the signaling mechanisms leading to type I IFN induction, future studies will fill in the remaining gaps in these signaling pathways. Further-more, we still have a lot to learn regarding the function of various cell types in IFN induction during in vivo infections, as well as the role type I IFNs play in host defense against different types of pathogens. In this review, we will discuss the recent progress made in elucidating the mechanisms controlling type I IFN production (Fig. 1)

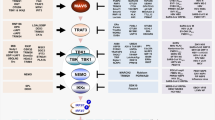
Pathways involved in activation of type I IFN production. Recognition of viral and bacterial components by host pattern recognition receptors (PRR) trigger signaling pathways that induce production of type I IFN. Viruses enter cells either by fusion at the plasma membrane or by endocytosis followed by fusion with the endosomal membrane, and entry into the cytoplasm. Viruses that reach the cytoplasmic compartment produce dsRNA during replication, which is recognized by the PRRs retinoic acid inducible gene-1 (RIG-1) and dsRNA-dependent protein kinase (PKR). Viruses that enter endocytic compartments are recognized by Toll-like receptors (TLR), TLR3, TLR7, TLR8, and TLR9. TLR9 recognizes CpG motifs of DNA viruses including murine cytomegalovirus (MCMV), herpes simplex virus (HSV) -1, and HSV-2. TLR7 and TLR8 recognize ssRNA from RNA viruses including vesicular stomatitis virus (VSV), and influenza virus. TLR3 recognizes dsRNA motifs of both types of viruses including MCMV. TLR4 is able to recognize the LPS component of Gram-negative bacteria, viral glycolipids, and the F protein of respiratory syncytial virus (RSV). Listeria monocytogenes (L. mono) enters cells by phagocytosis, and subsequently lyses the phagosomal membrane to escape into the cytoplasm. An unknown PRR recognizes intracellular L. mono. All of these PRRs can activate production of type I IFNs, likely through activation of the kinases TANK-binding kinase-1 (TBK1) and/or the inducible IκB kinase (IKK-i,). TLR9 and TLR7 utilize the adaptor protein myeloid differentiation factor-88 (MyD88), while TLR3 and TLR4 utilize Toll/IL-1 receptor domain-containing adaptor inducing IFNβ (TRIF) to transmit signals to TBK1/IKK-i. TBK1 and IKK-i activate the transcription factors interferon regulatory factor (IRF)-3 and IRF7, which induce the transcription of type I IFNs. Type I IFNs have pleiotropic effects and activate multiple components of host innate and adaptive immune responses.
Activation of type I IFN production by pattern recognition receptors
Pattern recognition receptors (PRRs) serve as first-line sentinels for innate immune detection of pathogenic infections, by recognizing various conserved molecular motifs termed “pathogen-associated molecular patterns” (PAMPs) 3. PRRs activate production of various cytokines and chemokines, allowing adaptation of the host environment for mounting an ideal immune response 3. The differences in the gene milieu induced by different PRRs may correlate to the type of invading pathogen and location of the infection, and assist in fine-tuning the immune response for specific pathogen clearance 3. Several PRRs, including members from a group of transmembrane signaling receptors known as Toll-like receptors (TLRs), as well as a few cytoplasmic PRRs, have been shown to activate production of pivotal cytokines in the innate immune defense system, the type I IFNs 3. Type I IFNs are responsible for inducing transcription of a large group of genes which play a role in host resistance to viral infections, as well as activating key components of the innate and adaptive immune systems including antigen presentation cells (APC) maturation and production of cytokines involved in activation of T cells, B cells, and natural killer (NK) cells 2.
Type I IFNs are transcriptionally regulated, and are induced following recognition of pathogen components during infection 4. In the classical positive feedback-production loop, IFNβ transcription is first activated by signals that induce cooperative binding of the transcription factors c-Jun/ATF-2, NF-κB, and interferon regulatory factor-3 (IRF3), to the IFNβ promoter 5. While NF-κB and c-Jun/ATF2 become activated by multiple types of stimuli and regulate transcription of large classes of genes, IRF3 is primarily under regulation for production of IFNβ 4, 5. IRF3 is constitutively expressed in all cells, and exists in a latent state in the cytoplasm of unstimulated cells 4. However, following stimulation with virus components including dsRNA or bacterial components including LPS, IRF3 becomes phosphorylated by the serine-threonine kinases TANK-binding kinase-1 (TBK1/T2K/NAK) or the inducible IκB kinase (IKK-i/IKKε) 6, 7, 8, 9, 10. IRF-3 then dimerizes, translocates into the nucleus, and combines with the coactivator CBP/P300 to activate the expression of IFNβ 4, 5. Following production, IFNβ initiates a positive feed-back loop by binding to IFNAR in an autocrine and paracrine manner 4, 5. IFNAR activates JAK protein tyrosine kinases (JAK1 and Tyk2) which phosphorylate STAT1 and STAT2 5. STAT1 and STAT2 together with IRF9 form a transcription factor complex termed IFN-stimulated gene factor 3 (ISGF3) 5. ISGF3 translocates into the nucleus and binds IFN-stimulated response elements (ISRE), to induce expression of a large group of “IFN-inducible” genes, including IRF7 and many others with anti-viral functions 4, 5. IRF7, a close relative of IRF3, is expressed primarily in lymphoid cells, and is transcriptionally induced in many cell types as a target of type I IFN signaling 11, 12. IRF7 also becomes activated by phosphorylation by TBK1 and/or IKK-i, and acts to further type I IFN expression by activating transcription of IFNα-non4 subtypes 10, 11.
TLR-mediated type I IFN production
In recent years, TLRs have been shown to recognize PAMPs from a broad array of pathogens and play key roles in subsequent activation of innate and adaptive immune responses 13. To date, 13 TLRs have been cloned in mammals (10 in human and 12 in mice), and each receptor seems to be involved in the recognition of a unique set of PAMPs from bacteria, viruses, fungi, and protozoa 13.
All TLRs contain extracellular leucine-rich repeat (LRR) domains which participate in ligand recognition, and intracellular TIR (Toll-interleukin (IL)-1 receptor) domains which transmit downstream signals via interactions with TIR domain-containing adaptor molecules including myeloid differentiation factor-88 (MyD88), TIR-associated protein (TIRAP/MAL), Toll/IL-1 receptor domain-containing adaptor inducing IFNβ (TRIF/TICAM-1), and Toll-receptor-associated molecule (TRAM/TIRP/TICAM-2) 14. All TLRs except TLR3 may utilize the adaptor molecule MyD88, to activate NF-κB and the mitogen-activated protein kinases (MAPKs) such as, extracellular signal-regulated kinase (ERK), p38, and c-Jun N-terminal kinase (JNK) 14. This MyD88-dependent pathway leads through the serine-threonine kinase IRAK, tumor necrosis factor (TNF) receptor-associated factor-6 (TRAF6), a MAPK kinase kinase (MAPKKK) termed transforming growth factor β-activated kinase (TAK-1), which results in activation of the IKK complex. The activated IKK complex can then lead to phosphorylation and subsequent degradation of IκB, the release of NF-κB, and activation of NF-κB-dependent genes, including the pro-inflammatory cytokines IL-1, IL-6 and TNFα 14. In addition, TAK-1 also can phosphorylate MKK3 and MKK6, two kinases upstream of p38 MAPK and JNK 14.
In addition to the MyD88-dependent signaling pathway that induces these inflammatory responses, the ability of individual TLRs to induce different signaling pathways has recently received much attention. The differential ability of TLRs to utilize specific TIR-adaptor molecules is thought to be the molecular basis for their inherent signaling differences 14. Several members of the TLR family have been shown to activate production of type I IFNs following ligand stimulation in various cell types 14. In particular, TLR3 and TLR4, which recognize viral dsRNA and Gram-negative bacterial LPS, respectively, can still induce certain signaling pathways, including type I IFN production, in MyD88-deficient cells 15, 16. The adaptor molecule TRIF mediates this MyD88-independent pathway leading to type I IFN production 17, 18, 19, 20, 21. In cells from TRIF knockout mice, TLR3 and TLR4-induced expression of IFNβ and subsequent IFN responses are defective 19, 20. Unlike TLR3, which can directly recruit TRIF, TLR4 uses the adaptor TRAM to transduce its signal to TRIF 18, 22, 23. Additionally, a MyD88-dependent pathway activated by TLR7, TLR8, and TLR9, leading to type I IFN production has been shown to occur in certain dendritic cell subtypes, particularly plasmacytoid dendritic cells (pDCs), also known as interferon-producing cells 24.
TLR4
TLR4 recognizes the lipid A or LPS component of the outer membrane of Gram-negative bacteria14. It is likely to be the major receptor contributing to type I IFN production in response to Gram-negative bacterial infections. In addition, TLR4 can also recognize various glycoproteins in viral envelopes. TLR4 was shown to recognize the fusion (F) protein of respiratory syncytial virus (RSV) and was required for viral clearance 25, 26. The envelope proteins (EnV) of the retroviruses murine mammary tumor virus (MMTV) and Moloney murine leukemia virus were both shown to interact with TLR4 27. However, whether or not TLR4 plays a major role in IFN production during viral infections remains to be determined.
Upon ligand recognition, the TLR4 complex can recruit multiple TIR-adaptor molecules including MyD88, TIRAP, TRIF, and TRAM 14. While MyD88 and TIRAP are mainly responsible for TLR4-mediated inflammatory responses, TRAM and TRIF signal to the kinase TBK1 for IRF3 activation and primary IFNβ production 14. Cells deficient in TRIF, TRAM, TBK1, or IRF3 have severe defects in TLR4-induced type I IFN production 7, 8, 9, 19, 20, 22, 28. TBK1 but not IKK-i may be the critical kinase involved in TLR4-induced IRF3 activation. We proposed that high levels of IKK-i expression in macrophages could compensate for a deficiency in TBK1 during viral infections, as IRF3 activation was not observably defective in TBK1−/− macrophages during Sendai virus infection 9. However, following LPS stimulation, a complete defect in IRF3 activation and IFN responses were seen in TBK1−/− macrophages and fibroblasts, but no defects were seen in IKK-i−/− macrophages, DCs, and fibroblasts 7, 8, 9.
TLR3
Studies have clearly shown that stimulation with dsRNA, a product commonly produced during viral replication stages, can induce type I IFN production through multiple pathways. TLR3 was shown to be required for full induction levels of pro-inflammatory cytokines and type I IFN in response to stimulation with exogenous polyI:C or dsRNA derived from the dsRNA virus, reovirus 29. The interaction of TLR3 with dsRNA is thought to occur in endocytic compartments 30.
However, the relevance of TLR3 in biological responses to viral infections is unclear, as TLR3-independent, cytoplasmic dsRNA recognition pathways exist and appear sufficient for host resistance to many viruses. Host responses to the DNA virus, murine cytomegalovirus (MCMV), have been shown to depend on TLR3, as TLR3−/−mice had higher viral titers and reduced production of pro-inflammatory cytokines and type I IFN 31. However, a separate study found no differences in T cell responses during MCMV infection between wild-type and TLR3−/−mice 32. Furthermore, measured responses to the RNA viruses including reovirus, Sendai virus, lymphocytic choriomeningitis virus (LCMV), Newcastle disease virus (NDV), vesicular stomatitis virus (VSV), and Influenza virus are unaffected in TLR3−/− mice 32, 33, 34, 35, 36. Interestingly, a recent study showed that TLR3−/− mice were actually more resistant than wild-type mice to death induced by intraperitoneally injected West Nile virus 37. Close examination revealed that despite a higher serum viral titer and reduced production of IL-6, TNFα, and IFNβ in TLR3−/− mice (indicating TLR3 does contribute to type I IFN responses during infection with this virus), these mice were better able to control viral entry into the brain and subsequent encephalitis-induced death 37. Production of TNFα was found to mediate blood-brain barrier leakiness, and thus the lower level of TNFα produced in TLR3−/− mice may mediate resistance seen in TLR3−/− mice 37. Furthermore, following direct CNS injection of virus, equal death rates were seen between wild-type and TLR3−/− mice 37. Thus, during infection with different types of viruses, the role of TLR3 appears to differ in its contribution to an effective immune response.
The TLR3-dependent pathway leads through TRIF, TBK1, and IRF3 to activate type I IFN production. Biochemical studies in support of this pathway have shown interactions between TLR3-TRIF, TRIF-TBK1, and TBK1-IRF3 6, 17, 21, 23, 38, 39, 40. Additionally, genetic studies in cells lacking these signaling components have shown similar degrees of defective IFN production in response to polyI:C 7, 8, 9, 19, 20, 22, 28. TRIF-deficient peritoneal macrophages can still activate IFNβ production in response to polyI:C, though in a much diminished and delayed fashion, similar to TLR3−/− peritoneal macrophages 20. TBK1 but not IKK-i may play a role in TLR3-induced IRF3 activation. IRF3 activation and IFNβ production were absent in TBK1−/− macrophages and fibroblasts exogenously stimulated with polyI:C, although IFNα and other IFN-inducible genes were still induced in a slightly delayed fashion 7, 9. Thus, further studies need to address the specific roles for TBK1 and IKK-i in TLR3 responses.
Recently, the PI3K-Akt pathway has been shown to function in IRF3 phosphorylation downstream of TLR3 41. PI3K could bind to TLR3 following stimulation with dsRNA 41. Interestingly, when PI3K activity was inhibited, although IRF3 was still phosphorylated at serine-396 and could localize to the nucleus, IRF3-dependant transcription was absent 41. This PI3K activity was shown to occur independently of TRIF 41. IRF3 is known to be phosphorylated at multiple sites, and 2D-gel analysis showed only partial phosphorylation of IRF3 occurred in the absence of PI3K activity, suggesting that the PI3K pathway is required for full activation of IRF3 41. However, the specific contributions of the TRIF and PI3K pathways to IRF3 phosphorylation, nuclear localization, and transcriptional activity remain to be elucidated.
The pathways leading from TLR3 to activation of IRF3 versus NF-κB diverge at TRIF. TRIF was shown to activate NF-κB separately through both its C-terminal and N-terminal domains 40. The N-terminal domain of TRIF was shown to interact independently with both TBK1 and TRAF6, and these two molecules may be in competition for binding to TRIF 40. A mutant TRIF lacking its TRAF6-binding motif was defective in NF-κB activation 39, 40. However, while TRAF6−/− fibroblasts were defective in activating TLR3-induced NF-κB, TRAF6−/− macrophages had no apparent defect in NF-κB activation in response to polyI:C 39, 42. The C-terminal of TRIF contains a receptor interacting protein (RIP) homotypic interaction motif (RHIM), and was shown to interact with the death domain-containing kinase RIP1 38, 43. Cells lacking RIP1 were defective in dsRNA-induced IκB phosphorylation and NF-κB-dependent gene transcription, but not IFNβ transcription 43. Thus, RIP1 may mediate one arm of TRIF-induced NF-κB activation.
Additional molecules suggested to play a role in dsRNA-induced type I IFN include the Rho GTPase Rac1 (ras-related C3 botulinum toxin substrate-1) 44. Rac1 was shown to be activated in cells stimulated with polyI:C or infected with influenza virus, and dominant-negative (DN) versions of Rac1 or the downstream activated kinase PAK1 (PI-3K/Rac1/p21-activated kinase-1), inhibited polyI:C or influenza virus-induced IRF3-reporter and IFNβ-reporter activities 44. Rac1-DN and PAK1-DN could also inhibit TBK1 and IKK-i activity 44. Furthermore, cells transfected with Rac1-DN had higher titers of Sendai and influenza viruses 44. Thus, the role of Rac1 and PAK1 in IFN induction pathways needs to be further explored. IKKβ, which can induce IRF3 phosphorylation in in vitro kinase assays, has also been suggested to play a role in TRIF-induced IRF3 activation in addition to NF-κB activation 38.
TLR7/8 and TLR9
TLR7 and TLR8 are the most closely related TLR family members, and trigger high levels of type I IFN production, particularly IFNα, in pDCs 36, 45, 46, 47, 48, 49. Human and murine TLR7 are able to recognize guanosine analogs and the imidazoquinoline compounds, imiquimod and R-848 50, 51, 52, 53. Human TLR8 can also recognize R-848, while TLR8 in mice is suggested to be non-functional 51, 52, 53. These low-molecular-weight compounds were previously known to have anti-tumor and anti-viral immunostimulatory activities, and imiquimod is used in clinical treatment of genital warts caused by human papillomavirus 54. The pathogen-associated ligand for TLR7 and TLR8 was later found to be G/U-rich ssRNA derived from viruses including human immunodeficiency virus (HIV) and influenza virus, although ssRNA of non-viral origin can also induce signaling through these receptors 45, 46. Infection with the ssRNA viruses, influenza virus and VSV, were also shown to induce IFNα production in pDCs through TLR7 and MyD88 36, 45, 49.
TLR9 recognizes unmethlyated CpG DNA motifs of bacteria and viruses, and TLR9 activates high levels of type I IFN production, particularly IFNα, in pDCs and bone-marrow derived DCs (BMDCs) 55, 56, 57, 58, 59, 60. Live infection with DNA viruses including HSV-1, HSV-2, and MCMV have been shown to induce high levels of type I IFN in pDCs through TLR9 56, 57, 58.
TLR7, TLR8, and TLR9 compose a subfamily of TLRs based on phylogenetic analyses, intracellular location, and signaling mechanisms 48. Recognition of molecular targets by TLR7/8 and TLR9 occurs in the endosome and signaling is inhibited by chloroquine, indicating the requirement for endosomal acidification in this process 36, 45, 48, 51, 53, 58. These TLRs exclusively use the adaptor MyD88 to transmit downstream signals including activation of type I IFN production 13. MyD88 can associate with and activate IRF7 but not with IRF3, leading to production of IFNα 61, 62. Recently, IRAK4 and TRAF6 were shown to be involved in MyD88-mediated activation of IRF7 61, 62. TRAF6 was shown to interact with IRF7 and the E3 ubiquitin ligase activity of TRAF6 was required for IRF7 activation 61, 62. However, the target of TRAF6-mediated ubiquitination remains to be defined. pDCs lacking IRAK4 were also deficient in TLR7 and TLR9-induced IFNα production 61. Additionally, in BMDCs, the ability of TLR7/8 and TLR9 ligands to induce IFNα production is not dependent on TBK1 62 (A. Perry and G. Cheng, unpublished observations). Thus, the question remains as to whether TBK1 and/or IKK-i are the relevant IRF7 kinases in the MyD88-IRF7 pathway. The cell-type specificity of TLR7, TLR8, and TLR9 in induction of type I IFN remains unclear. While high levels of type I IFN can be induced by these ligands in certain DC subsets, particularly pDCs, type I IFN production is not induced in macrophage cells and conventional DCs, which respond to these TLR ligands by producing pro-inflammatory cytokines 36, 46, 49, 56, 59, 63, 64, 65, 66, 67, 68. Thus, the differences in signaling components existing in these cell types need to be compared.
Type I IFN production induced by cytoplasmic receptors
PKR
Intracellular pathways that lead to type I IFN production in response to dsRNA have been shown to occur independently of TLR3. No defects are seen in type I IFN production in TLR3-knockout fibroblasts or BMDCs when polyI:C is introduced intracellularly via transfection 8, 64, 69. In BMDCs, transfection of polyI:C required the dsRNA-dependent protein kinase (PKR) to activate IFN production 64. PKR is a cytoplasmic serine-threonine kinase with two conserved dsRNA binding domains in its N-terminal region 70. PKR belongs to a class of more than 20 dsRNA binding proteins, which have been reported to interact with as little as 11 bp of dsRNA independent of the RNA sequence 70. PKR is activated by autophosphorylation following binding to dsRNA or phosphorylation by upstream kinases including PKR-activating protein (PACT), another dsRNA-binding kinase 70. Once activated, PKR phosphorylates the protein synthesis factor eIF-2α, which leads to inhibition of translation initiation and halts viral replication 70. PKR is also believed to play a role in NF-κB activation in response to dsRNA and viral infections 71, 72, 73. The potency of PKR in host anti-viral responses is highlighted by the numerous viruses that encode genes to inhibit PKR activity 74.
The role of PKR in type I IFN induction during viral infection remains complicated. PKR but not TLR3 was required for IFNα production in response to transfected polyI:C, while TLR3 was more important in response to exogenous polyI:C, indicating that the importance for each of these receptors depends on the subcellular location of dsRNA 34, 64, 72. However, IL-12 is still produced in response to intracellular polyI:C in PKR−/− cells 64. Thus, upon stimulation with dsRNA, PKR acts to enhance type I IFN production, while a PKR-independent mechanism exists to recognize intracellular dsRNA and activate proinflammatory cytokines. However, whether or not PKR signals lead to activation of IRF3 remains uncertain, and the contribution of PKR in induction of type I IFNs may lie solely in its ability to activate NF-κB. Furthermore, because PKR plays such an important role in limiting viral replication, it has been difficult to study IFN-induction in PKR-deficient cells infected with viruses. One study found a defect in type I IFN induction in PKR−/− cells infected with VSV 72. In contrast, no defects have been shown in IFN induction in PKR−/− cells infected with Sendai virus, influenza virus, and NDV 34, 47, 49, 75. However, this may be due to the ability of viruses to multiply to much higher titers in PKR−/− cells, and the large amounts of dsRNA or other viral PAMPs produced in PKR−/− cells may activate alternative IFN-induction pathways to an extent that it masks the requirement for PKR 76, 77. The influenza virus protein NS1 has been shown to inhibit PKR activation by sequestering dsRNA, and influenza virus lacking functional NS1 (delNS1) can strongly induce type I IFN production in various cell types 49, 64, 78, 79. This was proposed to be mediated by dsRNA sensors including PKR, although cells lacking PKR had no defect in IFN production during infection with delNS1 49. However, it was pointed out that NS1-deficient influenza viruses replicate to 105 higher titers in PKR−/− cells 64, 76. The vaccinia virus dsRNA-binding protein E3L which can bind to PKR and inhibit its activity, could also inhibit NDV-induced IRF3 phosphorylation 75. Thus, it remains unclear if PKR signaling leads to activation of IRF3.
RIG-1
Very recently, RIG-1 (retinoic acid inducible gene-1) was identified as a potential viral PRR able to recognize cytoplasmic dsRNA 35. RIG-1, a DexD/H box-containing RNA helicase, was found to activate IFNα/β production in response to transfected dsRNA and NDV infection 35. The RNA helicase domain was shown to interact with dsRNA, while overexpression of the caspase recruitment domain (CARD) of RIG-1 activated IRF3 and NF-κB 35. Dominant-negative RIG-1 inhibited NDV-induced IFN but not exogenous polyI:C or TLR3 signaling, providing further evidence for this intracellular pathway being separate from the TLR3 pathway 35. Overexpression of RIG-1 inhibited replication of VSV and encephalomyocarditis virus (ECMV) in L929 cells 35. However, this same study found no defects in IRF3 activation during NDV infection in fibroblasts deficient in TRIF or TBK1 35. This is in contrast to a study in which TBK1 was required in fibroblasts for NDV-induced IRF3 activation and IFN production 7.
Future studies need to address whether or not PKR and RIG-1 represent independent pathways of dsRNA recognition leading to type I IFN production, as well as to elucidate downstream signaling molecules leading to IRF3 and IRF7 activation. The signaling components downstream of RIG-1 for instance, may be CARD-domain-containing proteins, as CARD domains typically mediate homotypic interactions. So far, polyI:C transfection into TBK1-knockout and TBK1/IKK-i-double knockout fibroblasts revealed both TBK1 and IKK-i are required for the intracellular dsRNA-induced IFN pathway in fibroblasts, however whether TBK1 or IKK-i participate in PKR or RIG-1-pathways remains to be explored 8.
TBK1 and IKK-i as IRF3 and IRF7 Kinases
TBK1 and IKK-i were simultaneously identified as having the ability to phosphorylate both IRF3 and IRF7 6, 10. However, whether or not these kinases prefer either substrate is unknown. RNAi targeting either TBK1 or IKK-i inhibited Sendai virus and VSV-induced IRF3 transcriptional activity 6, 10. Thus, TBK1 and IKK-i may both be important in IFN induction during viral infections. In fibroblast cells, which express low levels of IKK-i, a lack of TBK1 resulted in defective IRF3 activation and IFN production in response to Sendai virus, VSV, and NDV 7, 8, 9, 69. The delayed IRF3 activation and IFN production seen in TBK1−/− fibroblasts may be attributed to virus-induced upregulation of IKK-i expression. Accordingly, reconstitution of TBK1−/− fibroblasts with IKK-i rescued the defects in IFN responses to Sendai virus 9. Macrophages express constitutively high levels of IKK-i, and no apparent defects could be seen in TBK1−/− macrophages infected with Sendai virus 9. Thus, it appears that TBK1 and IKK-i may be redundant in function when it comes to viral infection. However, the question remains as to why TBK1−/− macrophages have defective IRF3 activation and IFN induction in response to TLR3 and TLR4 ligands even though IKK-i is highly expressed in these cells 9. Additionally, the role of TBK1 and IKK-i in activation of IRF3 and IRF7 by other PRR pathways remains to be explored. Thus, do TBK1 and IKK-i play overlapping roles during viral infection, or do they mediate different pathways of viral recognition leading to type I IFN responses?
Type I IFN production in response to viral infections
Much progress in defining the components of PRR pathways leading to type I IFN production has allowed classification of viruses based on pathways they activate during infection (Tab. 1). Each virus is likely to be detected by multiple PRRs throughout their infection lifecycle, although certain PRRs may be more important for inducing production of type I IFNs. Different mechanisms of viral detection may be mediated by the subcellular location of viral patterns and mechanisms of cell entry by the virus. Viruses can enter cells by several different mechanisms. Non-enveloped viruses enter cells by binding to the cellular membrane and generating a pore or disrupting the membrane 80. Some enveloped viruses enter cells by fusion of the viral envelope at the plasma membrane and release of the viral particle into the cytoplasm 81. A more prominent mechanism for enveloped viruses is entry by endocytosis, in which viruses are taken up into endocytic vesicles via phagocytosis or pinocytosis, and subsequently enter the cytoplasm by endosomal membrane fusion or lysis of the endosomal membrane 81. Thus, endosomally-located PRRs including TLR3, TLR7/8, and TLR9, appear to be strategically located for detecting such viral invaders. However, it is not known if pDCs, which play the most prominent role in IFN production during viral infections, are actively infected by viruses or instead actively uptake and survey extracellular particles. Once the virus enters the host cytoplasm and begins replication, cytoplasmic detectors of viral patterns become more important, particularly in recognition of dsRNA, which is produced during replication of both DNA and RNA viruses. As virally encoded proteins that inhibit IFN responses now come into play, the host appears to have evolved multiple mechanisms to detect cytoplasmic products of viral replication, particularly dsRNA. Here we will discuss the host mechanisms of viral detection that lead to type I IFN production against several well studied viruses.
HSV-1 and HSV-2
HSV-1 and HSV-2 belong to the family of herpes-viruses, which are composed of four layers: a large dsDNA genome, enclosed in a caspid, surrounded by a tegument protein coat, and then by a glycoprotein-bearing lipid bilayer envelope 82. These herpesviruses primarily infect neurons and epithelial cells, although leukocytes can also be infected 82. Several envelope glycoproteins, particularly glycoprotein-D, mediate entry by binding to cellular receptors, triggering fusion with the plasma membrane and acess of the tegument-enclosed viral particle into the cytoplasm 82. Endocytosis may also occur in some cell types, perhaps pDCs, while viral particles present in endosomes are considered as primarily progeny viruses 82. Exit of progeny viruses from the cell requires envelopment by the golgi or trans-golgi network 82.
The host is equipped with multiple PRRs able to recognize HSV-1 and HSV-2 components and lead to immune cell activation. pDCs have emerged as the primary cell types producing type I IFN while infected by HSV-1 and HSV-2, via TLR9 recognition of viral DNA 58, 68. pDCs may come into contact with HSV via endocytosis of viral particles, as viral replication is not required for the IFN response 58, 68. IFNα induction can still occur in various TLR9−/− and MyD88−/− bone marrow cell subsets, although heat treatment of HSV-1 abrogated TLR9-independent responses, indicating recognition by other PRRs requires HSV replication or recognition of heat-labile molecules 83. For instance, PKR may become activated by dsRNA induced by replicating HSV, although it is not known if this leads to type I IFN production 84. Proinflammatory cytokine production in response to HSV can occur through an TLR9-dependent, TLR9-independent but MyD88-dependent way, as well as MyD88-independent mechanisms 83, 85. IL-6 production at least, may be mediated by activation of TLR2 by HSV-1 86. Furthermore, MyD88−/− and TLR9−/− mice infected with HSV-1 in the footpad or cornea were able to control viral replication equivalently to wild-type mice 68. Take together, the contribution of type I IFN production by pDCs to the host response remains to be explored.
MCMV
MCMV is a β-herpesvirus studied as a murine model for human CMV infection 87. β-herpesviruses are composed of large dsDNA genomes surrounded by 3 additional layers similar to the α-herpesviruses 88. MCMV can infect a range of cell types via fusion with the plasma membrane, but exhibit a tropism for hematopoietic cells 87. After infection, MCMV replicates to high levels in the liver and spleen, leading to liver dysfunction and immune suppression 87.
MCMV is detected by TLR9 and induces type I IFN and IL-12 production in DCs 31, 56. However, in vivo cytokine production still occurs in TLR9-knockout mice, indicating there must exist other PRRs to recognize MCMV 31, 56. TLR3 may recognize MCMV dsRNA as mice deficient in TLR3 had a decrease in IFNα/β production, increased MCMV titers, and a decrease in overall survival 31. Further evidence of a relevant role for the TLR3 pathway in MCMV recognition is seen in mice deficient in TRIF, which also shows defective MCMV clearance 19. Clearance of MCMV requires activation of NK cells, which detect MCMV-infected cells by the NK receptor Ly49H 87. Type I IFN is known to activate NK cells through inducing cytokines including IL-15 2. Thus, optimal host resistance to MCMV requires multiple PRRs that lead to cytokine production including type I IFNs, as well as the NK receptor LY49H.
Influenza virus and VSV
Similar mechanisms of host recognition have been shown for two negative-stranded ssRNA viruses of different families that enter cells by similar mechanisms. Influenza virus and VSV enter cells via receptor-mediated endocytosis followed by low pH-dependent fusion with the endosomal membrane and release of the viral ribonucleoprotein (vRNP) into the cytoplasm 81, 89. However, VSV gene expression occurs in the cytoplasm, while influenza virus vRNPs are imported into the nucleus for gene expression 81, 89. Both of these viruses have been shown to trigger type I IFN production via recognition of viral ssRNA by TLR7 in endosomes of pDCs in a replication-independent manner 36, 45, 49. Thus, dissolution of the viral particle in the endosome releases ssRNA to trigger TLR7-induced IFNα.
However, even though most type I IFN production induced by influenza virus is dependent on the TLR7-MyD88 pathway, survival following influenza virus infection was similar in wild-type and MyD88−/− mice 49. It was hypothesized that other PRRs may contribute to recognition of influenza and activation of an adequate immune response. Although influenza is a ssRNA virus, and would produce dsRNA during replicative steps, no defects were seen in IFNα production in TLR3−/− PBMCs infected with influenza virus 36. However, induction of IFNα by influenza virus was strongly enhanced in BMDCs if the virus lacked a functional NS1 protein, which acts to sequester dsRNA and inhibits activation of dsRNA sensors including PKR 49, 64, 79. Furthermore, this increase in IFN production was independent of MyD88, and required viral replication, indicating type I IFN production may be occurring as a result of dsRNA sensing by TLR3, PKR, or RIG-1, normally inhibited by NS1 49. However, PKR was not required for this response, indicating NS1 may inhibit multiple dsRNA recognition pathways 49. Thus, influenza virus infections are detected by TLR7, as well as dsRNA sensing pathways that can be inhibited by NS1.
VSV is also likely detected by dsRNA sensing pathways in addition to detection by TLR7. VSV has been shown to be able to trigger IRF3 activation and type I IFN production in a TBK-dependent, but TLR3 and TRIF-independent manner, indicating dsRNA signaling pathways besides TLR3 can utilize TBK1 but not TRIF to induce IFN production 8, 69. RIG-1 may be a candidate, as RIG-1 overexpressing cells had lower VSV titers 35.
Cell type specificity
While all cell types appear equipped with the ability to respond to viral infections by producing type I IFN, levels of production and IFN subtypes induced differ between various cell types. These differences are important in defining the roles that different cell types play in the host response to viral infections. Innate immune cells act as sentinels of viral invasion in a virus-replication-independent mechanism by TLRs, while non-immune cells which do not express high levels of TLRs can still produce type I IFNs in response to infection with replicating viruses or cytoplasmically-targeted dsRNA 45, 47, 58, 68. A rare DC subset, pDCs, have emerged as the major type I IFN producers during stimulation with TLR7/8 and TLR9 ligands and infection with viruses that activate signaling through these TLRs, including MCMV, HSV-2, VSV, and influenza virus 36, 45, 46, 56, 58, 64, 66, 68, 83, 90, 91, 92, 93, 94. The importance of these cells in IFN responses has been shown by lack of IFN production during MCMV infection in pDC-depleted mice 56, 66, 93. In pDCs, ligands for TLR7/8 and TLR9 upregulate high amounts of IFNα, while macrophages and conventional DCs respond to these ligands by activating NF-κB to produce proinflammatory cytokines including IL-12 but not much type I IFN 36, 46, 49, 56, 59, 63, 64, 65, 66, 67, 68. Thus, while both cell types are able to sense and respond to these ligands, downstream signaling outcomes are different. pDCs were also shown to be major producers of IFNα in response to transfected dsRNA and infection with the ssRNA virus RSV (which enters cells by fusion at the plasma membrane), in a replication-dependent manner 47. Thus, pDCs appear to be the major type I IFN producers even in response to viruses that may bypass TLR7/8 and TLR9 recognition in endocytic compartments 36, 47.
The molecular mechanisms governing the ability of pDCs to produce high levels of type I IFN is currently a focus of intense study. Expression levels of TLR signaling components has been shown to differ between pDCs and other cell types and may partly explain the differences in signaling outcomes 95. In mice and humans, pDCs express high levels of TLR7 and TLR9 compared to monocytes and conventional DCs although this does not explain the differences in downstream signaling outcomes 63, 67, 9596, 97. TLR7 and TLR9 activate type I IFN through a MyD88-IRF7 pathway, and relatively high constitutive expression levels of IRF7 are seen in pDCs 94, 97, 98. During VSV infection, which activates IFN production through TLR7 in pDCs, pDCs but not fibroblasts can upregulate early IFNα-non4 production independently of feedback signaling through IFNAR 36, 99. In contrast, in fibroblasts, early VSV-induced IFNα production is limited to the IRF3-regulated IFNα4 subtype 99. Thus, higher expression levels of IRF7 and other signaling components contribute to the ability of pDCs to produce high levels of type I IFN following TLR stimulation.
Differences in expression levels of IFN induction pathway members have also been observed between other cell types. For instance, the inducible kinase IKK-i is preferentially expressed in lymphoid cells although expression in specific cell subsets has not yet been investigated 100. The closely related kinase TBK1 however, seems to be constitutively expressed in most cell types 101. Infection of TBK1−/− fibroblasts but not TBK1−/− macrophages with Sendai virus resulted in defective IRF3 activation and type I IFN production, likely because IKK-i functions redundantly in macrophages 7, 8, 9. Thus, IKK-i expression may equip certain cell types with a greater ability to respond to virus infections.
Type I IFN production in response to bacterial infection
The first correctly described TLR4 ligand was the Lipid A component from LPS of Gram-negative bacteria. Stimulation with Gram-negative bacteria including Salmonella typhimurium and Escherichia coli or LPS derived from these bacteria induces type I IFN production 102, 103, 104, 105. Other bacterial components that induce type I IFN include bacterial DNA and Gram-negative flagellin, indicating other bacterial infections may also activate type I IFN production 60, 103.
Listeria monocytogenes (LM) is a Gram-positive facultative intracellular bacterium that is mainly contracted through consumption of contaminated foods. LM infections are primarily concerning in immunocompromised individuals where sepsis and meningitis may develop, and in pregnant women where the fetus may become infected and lead to septic abortion 106. LM infects and resides in macrophages, as well as intestinal epithelial cells and hepatocytes by inducing phagocytosis 106. After phagosomal uptake, LM secretes listeriolysin O (LLO), a hemolysin, to form pores in the phagosomal membrane and allow cytoplasmic translocation 106. Thereafter, LM mobilizes host actin filaments to propel itself through the host cell cytoplasm and penetrate directly into neighboring cells without becoming exposed to the extracellular space 106.
In vitro and in vivo infection studies have shown that LM can induce production of type I IFN 107, 108, 109, 110, 111, 112, 113, 114, 115. While LM infection can induce TLR and MyD88-dependent NF-κB activation and production of proinflammatory cytokines including TNFα and IFNγ, induction of type I IFN is independent of TLR9, TLR2, TLR4, MyD88, TRIF, and TRAM, and occurs at a later time point than NF-κB activation 112, 113, 114, 115. Instead, LM-induced type I IFN production occurs through a pathway dependent on TBK1, IRF3, and primary induction of IFNβ 113, 114, 115. Further analyses revealed the likelihood of an intracellular receptor mediating LM induced type I IFN, as induction kinetics corresponded with phagosomal escape, and induction only occurred if LM encoded LLO, allowing the bacterium to escape into the cytoplasm of the host cell 108, 110, 112, 113, 114. The NOD (nucleotide-binding oligomerization domain) family of intracellular PRRs do not appear to be involved in LM-induced type I IFN, as there were no defects in IFN induction in macrophages lacking NOD2 or the NOD1/2 adaptor molecule RIP2 114, 115. Thus, LM appears to activate type I IFN production through a unique pathway that is independent of TLRs, but converges at the IRF3 kinase TBK1.
Contrary to what one would first assume, the production of type I IFN during LM infections was actually detrimental to the host 109, 110, 111, 113, 115. IRF3−/− or IFNAR−/− mice which are unable to either produce or respond to type I IFNs, were more resistant to LM, having greater survival rates and lower LM titers in the liver and spleen than wild-type mice 109, 110, 111, 113. Additionally, injection of mice with polyI:C enhanced LM-induced death in wild type but not IFNAR−/− mice, further supporting that the response to type I IFNs is detrimental to the host during LM infection 113. In this setting, type I IFN production appears to sensitize infected splenocytes to LM-induced apoptosis. This increase in splenocyte apoptosis was accompanied by IFN-induction of proapoptotic gene programs including PKR, the death-receptor ligand TRAIL, and Daxx 109, 111, 112, 113. Mice deficient in TRAIL were also more resistant to LM-induced splenocyte apoptosis and had lower spleen and liver LM titers 116.
Infection with the intracellular Gram-positive pathogen Mycobacterium tuberculosis (Mtb), the causative agent of tuberculosis, has also been shown to induce production of type I IFNs. Llike LM, Mtb infects and resides in host macrophages. Infection of various cell types with Mtb has been shown to activate NF-κB and IRF-3, and induce expression of both IFNβ and IFNα 117, 118, 119, 120. A hypervirulent strain of Mtb that induced higher levels of IFNα than another virulent strain, was shown to kill infected mice at a faster rate 120. Although production of Th1 cytokines was lower in mice infected with the hypervirulent strain, co-administration of IFNg with infection had no effects on survival, while co-administration of IFNα/β with infection further enhanced killing by the Mtb strain 120. Thus, although early death induced by this Mtb strain may be partially due to the failure to elicit proper Th1 responses, production of type I IFN greatly enhanced death 120. A related pathogen, Mycobacterium bovis, was shown to have enhanced replication rates in macrophages pre-treated with type I IFNs 121. Thus, in contrast to the critical role played by type I IFNs in host anti-viral responses, production of these cytokines during infection with intracellular Gram-positive bacterial pathogens appears to be detrimental to the host.
Conclusion
Type I IFNs are rapidly induced and play an essential role in defense against viral infection at any stage of the virus life cycle 122. By feeding back in an autocrine and paracrine manner, IFNα/β induces an anti-viral state in surrounding cells by inducing transcription of genes involved in apoptosis, anti-growth, and innate and adaptive immune cell activation. Some of the most well-studied anti-viral genes induced by type I IFNs include PKR, ADAR (adenosine deaminase acting on RNA), OAS (2',5'-oligoadenylate synthetase), RNase L, and Mx proteins. PKR and ADAR lead to inhibition of translation initiation and RNA editing, respectively. OAS is a dsRNA-dependent synthetase that activates the endoribonuclease RNase L to degrade ssRNA 123. For example, activated RNase L can degrade respiratory syncytial virus M2 RNA 124. Mx proteins are dynamin superfamily GTPases that act on a wide range of viruses by binding target viral proteins to inhibit virus replication. For example, MxA can bind to the nucleocapsid of Thogoto virus and block nuclear import 125.
Besides inducing proteins with direct anti-viral effects, type I IFNs regulate multiple aspects of innate and adaptive immunity. Type I IFN regulates the function of NK cells by inducing production of IL-15 to promote NK cell survival and proliferation 126. Type I IFN production has been shown to stimulate DC maturation by upregulating surface expression of MHC and co-stimulatory molecules CD80, CD86, and CD40 2. Type I IFN may also induce differentiation of pDC subsets into mature APCs 66, 127. Type I IFN plays a role in activation of naïve CD8+ T cells, survival of activated CD4+ and CD8+ cells, and development and proliferation of B cells 2. Activated IRF3 directly activates production of the chemokines IP-10 (IFN-γ inducible protein 10, CXCL10) which recruits lymphocytes and monocytes, and RANTES (regulated on activation, normal T cell expressed and secreted, CCL5) which attracts activated and memory T cells, eosinophils and basophils 4, 128, 129.
Due to the vast effects that type I IFNs have on stimulating and shaping the immune response, it is no wonder that many studies have found indispensable roles for type I IFN in anti-viral host responses. Most, if not all, virus infections are recognized by host PRRs and lead to type I IFN production. The collective recent studies have shown that multiple TLRs including TLR3, TLR4, TLR7, TLR8, and TLR9, as well as several intracellular PRRs including RIG-I and PKR are involved in type I IFN production upon recognition of viruses. The activation of PRRs triggers complex signaling pathways which induce numerous effective proteins that play important roles in host responses to pathogen invasion. A single virus can be recognized by multiple PRRs through different viral patterns. While this may have evolved as a response to viral-inhibition of IFN responses, activation of different PRRs in different cell types leads to induction of different gene programs and subsequent immune responses. How the role for production of type I IFN differs under these various conditions remains to be studied.
At the same time, many viruses have developed the ability to escape or inhibit the function of IFNs 74. Many viral products can block the IFN associated signal, such as the influenza NS1 protein and vaccinia virus E3L protein which inhibit PKR, and vaccinia virus-encoded soluble IFN-α/β receptor decoys 74, 130. The anti-IFN ability of viruses is reflected in some problems of clinical therapy. Type I IFN is the primary antiviral therapeutic used against hepatitis C virus (HCV) 74. However, HCV resistance to IFN treatment is prominent, likely due to mutations in the NS5A protein which acts to inhibit PKR and JAK-STAT signaling 74.
While type I IFNs play a critical role in host resistance to many types of viruses, it is very interesting that type I IFN production is also important to the host during infection with intracellular bacterial pathogens. Production of type I IFN during LM infection sensitized macrophages and lymphocytes to cell death 109, 111, 113. Type I IFN production may also be detrimental to the host during infection of Mtb 120. Thus, the question remains as to whether this response is true for all bacteria, for all intracellular bacteria, or just a few specific pathogens. Both Mtb and LM primarily infect macrophages, and it was also this cell type that exhibited cell death during LM infection 109, 113. Thus, is this response specific to macrophages as the host cell, or are other cell types also sensitized to IFN-mediated cell death? The host receptor recognizing the presence of intracellular LM has not yet been identified and it is interesting to note that LM may activate a completely unique pathway. Thus, has the host evolved a unique pathway to produce type I IFN upon recognition of cytoplasmic bacteria such as LM, or has the bacterium evolved to induce type I IFN production for its own benefit? Furthermore, studies need to address the role for type I IFNs in the host response to acute versus chronic infections. High dosages of LM were used to demonstrate acute infections in these experiments, and in such scenarios it may be true that overproduction of type I IFNs may be detrimental to the host. During infections with lower doses of LM, would this same effect be observed, or would type I IFN production benefit the host as it does during viral infections? Immunized IFNAR−/− mice were able to mount effective memory responses similar to that of wild-type mice during a secondary challenge with LM 110. Thus, type I IFN may not inhibit activation of adaptive immune responses (mainly CD8+ T cells) required for the clearance of LM. However, the effect of type I IFN on aspects of immune cell activation during LM infection needs to be further clarified.
Much progress has been made in understanding the induction and function of type I IFNs. Type I IFN and inducers of type I IFN including the TLR7 ligand imiquimod, are used clinically for treatment of virus originated diseases including hepatitis B, hepatitis C, multiple sclerosis, and genital warts caused by human papillomavirus 54, 131. IFNα is also a useful and active agent in clinical oncology for treatment of several malignant disorders including myeloma, renal cell carcinoma, and melanoma 132. However, production of type I IFN during certain intracellular bacterial infections including LM and Mtb has detrimental effects on the host. Type I IFN has also been associated with enhanced pathogenesis of autoimmune diseases including systemic lupus erythematosus and type I diabetes 2. Since type I IFNs play both positive and negative roles in a variety of immunological diseases, it is important to understand their mechanism of induction and action, in order to harness these powerful products of nature for our benefit.
Abbreviations
- BMDC:
-
bone marrow-derived dendritic cell
- DN:
-
dominant-negative
- ECMV:
-
encephalomyocarditis virus
- HSV:
-
herpes simplex virus
- IFN:
-
interferon
- IFNAR:
-
type I interferon α/β receptor
- IKK-i:
-
inducible IκB kinase
- IRF3:
-
interferon regulatory factor-3
- ISGF3:
-
IFN-stimulated gene factor-3
- ISRE:
-
IFN-stimulated response elements
- LCMV:
-
lymphocytic choriomeningitis virus
- LM:
-
Listeria monocytogenes
- LRR:
-
leucine-rich repeat
- MCMV:
-
murine cytomegalovirus
- Mtb:
-
Mycobacterium tuberculosis
- MyD88:
-
myeloid differentiation factor-88
- NDV:
-
Newcastle disease virus
- NOD:
-
nucleotide-binding oligomerization domain
- PAK1:
-
PI-3K/Rac1/p21-activated kinase-1
- PAMP:
-
pathogen-associated molecular patterns
- pDC:
-
plasmacytoid dendritic cell
- PKR:
-
dsRNA-dependent protein kinase
- PRR:
-
pattern recognition receptor
- Rac1:
-
ras-related C3 botulinum toxin substrate-1
- RIG-1:
-
retinoic acid inducible gene-1
- RIP:
-
receptor interacting protein
- RSV:
-
respiratory syncytial virus
- TBK1:
-
TANK-binding kinase-1
- TIR:
-
Toll-interleukin-1 receptor
- TIRAP:
-
TIR-associated protein
- TLR:
-
Toll-like receptor
- TRAF6:
-
tumor necrosis factor receptor-associated factor-6
- TRAM:
-
Toll-receptor-associated molecule
- TRIF:
-
Toll/IL-1 receptor domain-containing adaptor inducing IFNβ
- VSV:
-
vesicular stomatitis virus
References
Isaacs A, Lindenmann J . Virus interference. I. The interferon. Proc R Soc Lond B Biol Sci 1957; 147:258–67.
Theofilopoulos AN, Baccala R, Beutler B, Kono DH . TypeI interferons (α/β) in immunity and autoimmunity. Annu Rev Immunol 2005; 23:307–35.
Iwasaki A, Medzhitov R . Toll-like receptor control of the adaptive immune responses. Nat Immunol 2004; 5:987–95.
Taniguchi T, Takaoka A . The interferon-α/β system in antiviral responses: a multimodal machinery of gene regulation by the IRF family of transcription factors. Curr Opin Immunol 2002; 14:111–6.
Malmgaard L . Induction and regulation of IFNs during viral infections. J Interferon Cytokine Res 2004; 24:439–54.
Fitzgerald KA, McWhirter SM, Faia KL, et al. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol 2003; 4:491–6.
McWhirter SM, Fitzgerald KA, Rosains J, et al. IFN-regulatory factor 3-dependent gene expression is defective in Tbk1-deficient mouse embryonic fibroblasts. Proc Natl Acad Sci U S A 2004; 101:233–8.
Hemmi H, Takeuchi O, Sato S, et al. The roles of two IκB kinase-related kinases in lipopolysaccharide and double stranded RNA signaling and viral infection. J Exp Med 2004; 199:1641–50.
Perry AK, Chow EK, Goodnough JB, et al. Differential requirement for TANK-binding kinase-1 in type I interferon responses to toll-like receptor activation and viral infection. J Exp Med 2004; 199:1651–8.
Sharma S, tenOever BR, Grandvaux N, et al. Triggering the interferon antiviral response through an IKK-related pathway. Science 2003; 300:1148–51.
Barnes B, Lubyova B, Pitha PM . On the role of IRF in host defense. J Interferon Cytokine Res 2002; 22:59–71.
Taniguchi T, Ogasawara K, Takaoka A, Tanaka N . IRF family of transcription factors as regulators of host defense. Annu Rev Immunol 2001; 19:623–55.
Beutler B . Inferences, questions and possibilities in Toll-like receptor signalling. Nature 2004; 430:257–63.
Akira S, Takeda K . Toll-like receptor signalling. Nat Rev Immunol 2004; 4:499–511.
Kawai T, Takeuchi O, Fujita T, et al. Lipopolysaccharide stimulates the MyD88-independent pathway and results in activation of IFN-regulatory factor 3 and the expression of a subset of lipopolysaccharide-inducible genes. J Immunol 2001; 167:5887–94.
Doyle S, Vaidya S, O'Connell R, et al. IRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity 2002; 17:251–63.
Yamamoto M, Sato S, Mori K, et al. Cutting edge: a novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-β promoter in the Toll-like receptor signaling. J Immunol 2002; 169:6668–72.
Fitzgerald KA, Rowe DC, Barnes BJ, et al. LPS-TLR4 signaling to IRF-3/7 and NF-κB involves the toll Adapters TRAM and TRIF. J Exp Med 2003; 198:1043–55
Hoebe K, Du X, Georgel P, et al. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature 2003; 424:743–8.
Yamamoto M, Sato S, Hemmi H, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 2003; 301:640–3.
Oshiumi H, Matsumoto M, Funami K, et al. TICAM-1, an adaptor molecule that participates in Toll-like receptor 3-mediated interferon-β induction. Nat Immunol 2003; 4:161–7.
Yamamoto M, Sato S, Hemmi H, et al. TRAM is specifically involved in the Toll-like receptor 4-mediated MyD88-independent signaling pathway. Nat Immunol 2003; 4:1144–50
Oshiumi H, Sasai M, Shida K, et al. TIR-containing adapter molecule (TICAM)-2, a bridging adapter recruiting to toll-like receptor 4 TICAM-1 that induces interferon-β. J Biol Chem 2003; 278:49751–62.
Liu YJ . IPC: Professional Type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu Rev Immunol 2005; 23:275–306.
Haynes LM, Moore DD, Kurt-Jones EA, et al. Involvement of toll-like receptor 4 in innate immunity to respiratory syncytial virus. J Virol 2001; 75:10730–7.
Kurt-Jones EA, Popova L, Kwinn L, et al. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat Immunol 2000; 1:398–401.
Rassa JC, Meyers JL, Zhang Y, et al. Murine retroviruses activate B cells via interaction with toll-like receptor 4. Proc Natl Acad Sci U S A 2002; 99: 2281–6.
Sakaguchi S, Negishi H, Asagiri M, et al. Essential role of IRF-3 in lipopolysaccharide-induced interferon-β gene expression and endotoxin shock. Biochem Biophys Res Commun 2003; 306:860–6.
Alexopoulou L, Holt AC, Medzhitov R, Flavell RA . Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature 2001; 413:732–8.
Matsumoto M, Funami K, Tanabe M, et al. Subcellular localization of Toll-like receptor 3 in human dendritic cells. J Immunol 2003; 171:3154–62.
Tabeta K, Georgel P, Janssen E, et al. Toll-like receptors 9 and 3 as essential components of innate immune defense against mouse cytomegalovirus infection. Proc Natl Acad Sci U S A 2004; 101:3516–21.
Edelmann KH, Richardson-Burns S, Alexopoulou L, et al. Does Toll-like receptor 3 play a biological role in virus infections? Virology 2004; 322:231–8.
Lopez CB, Moltedo B, Alexopoulou L, et al. TLR-independent induction of dendritic cell maturation and adaptive immunity by negative-strand RNA viruses. J Immunol 2004; 173:6882–9.
Honda K, Sakaguchi S, Nakajima C, et al. Selective contribution of IFN-α/β signaling to the maturation of dendritic cells induced by double-stranded RNA or viral infection. Proc Natl Acad Sci U S A 2003; 100:10872–7.
Yoneyama M, Kikuchi M, Natsukawa T, et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol 2004; 5:730–7.
Lund JM, Alexopoulou L, Sato A, et al. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc Natl Acad Sci U S A 2004; 101:5598–603.
Wang T, Town T, Alexopoulou L, et al. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat Med 2004; 10:1366–73.
Han KJ, Su X, Xu LG, et al. Mechanisms of the TRIF-induced interferon-stimulated response element and NF-κB activation and apoptosis pathways. J Biol Chem 2004; 279:15652–61.
Jiang Z, Mak TW, Sen G, Li X . Toll-like receptor 3-mediated activation of NF-kappaB and IRF3 diverges at Toll-IL-1 receptor domain-containing adapter inducing IFN-β. Proc Natl Acad Sci U S A 2004; 101:3533–8.
Sato S, Sugiyama M, Yamamoto M, et al. Toll/IL-1 receptor domain-containing adaptor inducing IFN-β (TRIF) associates with TNF receptor-associated factor 6 and TANK-binding kinase 1, and activates two distinct transcription factors, NF-kappaB and IFN-regulatory factor-3, in the Toll-like receptor signaling. J Immunol 2003; 171:4304–10.
Sarkar SN, Peters KL, Elco CP, et al. Novel roles of TLR3 tyrosine phosphorylation and PI3 kinase in double-stranded RNA signaling. Nat Struct Mol Biol 2004; 11:1060–7.
Gohda J, Matsumura T, Inoue J . Cutting edge: TNFR-associated factor (TRAF) 6 is essential for MyD88-dependent pathway but not toll/IL-1 receptor domain-containing adaptor-inducing IFN-β (TRIF)-dependent pathway in TLR signaling. J Immunol 2004; 173:2913–7.
Meylan E, Burns K, Hofmann K, et al. RIP1 is an essential mediator of Toll-like receptor 3-induced NF-kappa B activation. Nat Immunol 2004; 5: 503–7.
Ehrhardt C, Kardinal C, Wurzer WJ, et al. Rac1 and PAK1 are upstream of IKK-epsilon and TBK-1 in the viral activation of interferon regulatory factor-3. FEBS Lett 2004; 567:230–8.
Diebold SS, Kaisho T, Hemmi H, et al. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 2004; 303:1529–31.
Heil F, Hemmi H, Hochrein H, et al. Species-specific recognition of single-stranded RNA via Toll-like receptor 7 and 8. Science 2004; 303:1526–9
Hornung V, Schlender J, Guenthner-Biller M, et al. Replication-dependent potent IFN-α induction in human plasmacytoid dendritic cells by a single-stranded RNA virus. J Immunol 2004; 173:5935–43.
Crozat K, Beutler B . TLR7: A new sensor of viral infection. Proc Natl Acad Sci U S A 2004; 101:6835–6.
Barchet W, Krug A, Cella M, et al. Dendritic cells respond to influenza virus through TLR7- and PKR-independent pathways. Eur J Immunol 2005; 35:236–42.
Hemmi H, Kaisho T, Takeuchi O, et al. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat Immunol 2002; 3:196–200.
Lee J, Chuang TH, Redecke V, et al. Molecular basis for the immunostimulatory activity of guanine nucleoside analogs: activation of Toll-like receptor 7. Proc Natl Acad Sci U S A 2003; 100:6646–51.
Jurk M, Heil F, Vollmer J, et al. Human TLR7 or TLR8 independently confer responsiveness to the antiviral compound R-848. Nat Immunol 2002; 3:499.
Heil F, Ahmad-Nejad P, Hemmi H, et al. The Toll-like receptor 7 (TLR7)-specific stimulus loxoribine uncovers a strong relationship within the TLR7, 8 and 9 subfamily. Eur J Immunol 2003; 33:2987–97.
von Krogh G, Lacey CJ, Gross G, et al. European course on HPV associated pathology: guidelines for primary care physicians for the diagnosis and management of anogenital warts. Sex Transm Infect 2000; 76:162–8.
Hemmi H, Takeuchi O, Kawai T, et al. A Toll-like receptor recognizes bacterial DNA. Nature 2000; 408:740–5.
Krug A, French AR, Barchet W, et al. TLR9-dependent recognition of MCMV by IPC and DC generates coordinated cytokine responses that activate antiviral NK cell function. Immunity 2004; 21:107–19.
Krug A, Luker GD, Barchet W, et al. Herpes simplex virus type 1 (HSV-1) activates murine natural interferon-producing cells (IPC) through Toll-like receptor 9. Blood 2004; 103:1433–7.
Lund J, Sato A, Akira S, et al. Toll-like receptor 9-mediated recognition of Herpes simplex virus-2 by plasmacytoid dendritic cells. J Exp Med 2003; 198:513–20.
Bauer M, Redecke V, Ellwart JW, et al. Bacterial CpG-DNA triggers activation and maturation of human CD11c-, CD123+ dendritic cells. J Immunol 2001; 166:5000–7.
Krieg AM . CpG motifs in bacterial DNA and their immune effects. Annu Rev Immunol 2002; 20:709–60.
Honda K, Yanai H, Mizutani T, et al. Role of a transductional-transcriptional processor complex involving MyD88 and IRF-7 in Toll-like receptor signaling. Proc Natl Acad Sci U S A 2004; 101:15416–21.
Kawai T, Sato S, Ishii KJ, et al. Interferon-α induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat Immunol 2004; 5:1061–8.
Ito T, Amakawa R, Kaisho T, et al. Interferon-α and interleukin-12 are induced differentially by Toll-like receptor 7 ligands in human blood dendritic cell subsets. J Exp Med 2002; 195:1507–12.
Diebold SS, Montoya M, Unger H, et al. Viral infection switches non-plasmacytoid dendritic cells into high interferon producers. Nature 2003; 424:324–8.
Doyle S, Vaidya S, O'Connell R, et al. IRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity 2002; 17:251–63
Asselin-Paturel C, Boonstra A, Dalod M, et al. Mouse type I IFN-producing cells are immature APCs with plasmacytoid morphology. Nat Immunol 2001; 2:1144–50.
Kadowaki N, Ho S, Antonenko S, et al. Subsets of human dendritic cell precursors express different toll-like receptors and respond to different microbial antigens. J Exp Med 2001; 194:863–9.
Krug A, Luker GD, Barchet W, et al. Herpes Simplex Virus type 1 (HSV-1) activates murine Natural Interferon-Producing cells (IPC) through Toll-Like Receptor 9. Blood 2004; 103:1433–7
tenOever BR, Sharma S, Zou W, et al. Activation of TBK1 and IKKvarepsilon kinases by vesicular stomatitis virus infection and the role of viral ribonucleoprotein in the development of interferon antiviral immunity. J Virol 2004; 78:10636–49.
Saunders LR, Barber GN . The dsRNA binding protein family: critical roles, diverse cellular functions. Faseb J 2003; 17:961–83.
Yang YL, Reis LF, Pavlovic J, et al. Deficient signaling in mice devoid of double-stranded RNA-dependent protein kinase. Embo J 1995; 14:6095–106.
Chu WM, Ostertag D, Li ZW, et al. JNK2 and IKKβ are required for activating the innate response to viral infection. Immunity 1999; 11:721–31.
Kumar A, Yang YL, Flati V, et al. Deficient cytokine signaling in mouse embryo fibroblasts with a targeted deletion in the PKR gene: role of IRF-1 and NF-kappaB. Embo J 1997; 16:406–16.
Katze MG, He Y, Gale M, Jr . Viruses and interferon: a fight for supremacy. Nat Rev Immunol 2002; 2:675–87.
Smith EJ, Marie I, Prakash A, et al. IRF3 and IRF7 phosphorylation in virus-infected cells does not require double-stranded RNA-dependent protein kinase R or Ikappa B kinase but is blocked by Vaccinia virus E3L protein. J Biol Chem 2001; 276:8951–7.
Bergmann M, Garcia-Sastre A, Carnero E, et al. Influenza virus NS1 protein counteracts PKR-mediated inhibition of replication. J Virol 2000; 74:6203–6.
Balachandran S, Roberts PC, Brown LE, et al. Essential role for the dsRNA-dependent protein kinase PKR in innate immunity to viral infection. Immunity 2000; 13:129–41.
Wang X, Li M, Zheng H, et al. Influenza A virus NS1 protein prevents activation of NF-kappaB and induction of α/β interferon. Dec. J Virol 2000; 74: 11566–73.
Hatada E, Saito S, Fukuda R . Mutant influenza viruses with a defective NS1 protein cannot block the activation of PKR in infected cells. J Virol 1999; 73:2425–33.
Hogle JM . Poliovirus cell entry: common structural themes in viral cell entry pathways. Annu Rev Microbiol 2002; 56:677–702.
Lakadamyali M, Rust MJ, Zhuang X . Endocytosis of influenza viruses. Microbes Infect 2004; 6:929–36.
Garner JA . Herpes simplex virion entry into and intracellular transport within mammalian cells. Adv Drug Deliv Rev 2003; 55:1497–513.
Hochrein H, Schlatter B, O'Keeffe M, et al. Herpes simplex virus type-1 induces IFN-α production via Toll-like receptor 9-dependent and -independent pathways. Proc Natl Acad Sci U S A 2004; 101:11416–21.
Taddeo B, Luo TR, Zhang W, Roizman B . Activation of NF-kappaB in cells productively infected with HSV-1 depends on activated protein kinase R and plays no apparent role in blocking apoptosis. Proc Natl Acad Sci U S A 2003; 100:12408–13.
Sato A, Iwasaki A . Induction of antiviral immunity requires Toll-like receptor signaling in both stromal and dendritic cell compartments. Proc Natl Acad Sci U S A 2004; 101:16274–9.
Kurt-Jones EA, Chan M, Zhou S, et al. Herpes simplex virus 1 interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proc Natl Acad Sci U S A 2004; 101:1315–20.
Krmpotic A, Bubic I, Polic B, et al. Pathogenesis of murine cytomegalovirus infection. Microbes Infect 2003; 5:1263–77.
Spear PG . Herpes simplex virus: receptors and ligands for cell entry. Cell Microbiol 2004; 6:401–10.
Letchworth GJ, Rodriguez LL, Del cbarrera J . Vesicular stomatitis. Vet J 1999; 157:239–60.
Siegal FP, Kadowaki N, Shodell M, et al. The nature of the principal type 1 interferon-producing cells in human blood. Science 1999; 284:1835–7.
Cella M, Jarrossay D, Facchetti F, et al. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat Med 1999; 5:919–23.
Kadowaki N, Antonenko S, Liu YJ . Distinct CpG DNA and polyinosinic-polycytidylic acid double-stranded RNA, respectively, stimulate CD11c- type 2 dendritic cell precursors and CD11c+ dendritic cells to produce type I IFN. J Immunol 2001; 166:2291–5.
Dalod M, Salazar-Mather TP, Malmgaard L, et al. Interferon α/β and interleukin 12 responses to viral infections: pathways regulating dendritic cell cytokine expression in vivo. J Exp Med 2002; 195:517–28.
Coccia EM, Severa M, Giacomini E, et al. Viral infection and Toll-like receptor agonists induce a differential expression of type I and lambda interferons in human plasmacytoid and monocyte-derived dendritic cells. Eur J Immunol 2004; 34:796–805.
Reis e Sousa C . Toll-like receptors and dendritic cells: for whom the bug tolls. Semin Immunol 2004; 16:27–34.
Brawand P, Fitzpatrick DR, Greenfield BW, et al. Murine plasmacytoid pre-dendritic cells generated from Flt3 ligand-supplemented bone marrow cultures are immature APCs. J Immunol 2002; 169:6711–9.
Dai J, Megjugorac NJ, Amrute SB, Fitzgerald-Bocarsly P . Regulation of IFN regulatory factor-7 and IFN-α production by enveloped virus and lipopolysaccharide in human plasmacytoid dendritic cells. J Immunol 2004; 173:1535–48.
Izaguirre A, Barnes BJ, Amrute S, et al. Comparative analysis of IRF and IFN-α expression in human plasmacytoid and monocyte-derived dendritic cells. J Leukoc Biol 2003; 74:1125–38.
Barchet W, Cella M, Odermatt B, et al. Virus-induced interferon α production by a dendritic cell subset in the absence of feedback signaling in vivo. J Exp Med 2002; 195:507–16.
Shimada T, Kawai T, Takeda K, et al. IKK-i, a novel lipopolysaccharide-inducible kinase that is related to IκB kinases. Int Immunol 1999; 11:1357–62.
Tojima Y, Fujimoto A, Delhase M, et al. NAK is an IkappaB kinase-activating kinase. Nature 2000; 404:778–82.
Utaisincharoen P, Anuntagool N, Limposuwan K, et al. Involvement of β interferon in enhancing inducible nitric oxide synthase production and antimicrobial activity of Burkholderia pseudomallei-infected macrophages. Infect Immun 2003; 71:3053–7.
Sing A, Merlin T, Knopf HP, et al. Bacterial induction of β interferon in mice is a function of the lipopolysaccharide component. Infect Immun 2000; 68:1600–7.
Eloranta ML, Sandberg K, Ricciardi-Castagnoli P, et al. Production of interferon-α/β by murine dendritic cell lines stimulated by virus and bacteria. Scand J Immunol 1997; 46:235–41.
Yaegashi Y, Nielsen P, Sing A, et al. Interferon β, a cofactor in the interferon gamma production induced by gram-negative bacteria in mice. J Exp Med 1995; 181:953–60.
Pamer EG . Immune responses to Listeria monocytogenes. Nat Rev Immunol 2004; 4:812–23.
Havell EA . Augmented induction of interferons during Listeria monocytogenes infection. J Infect Dis 1986; 153:960–9.
O'Riordan M, Yi CH, Gonzales R, et al. Innate recognition of bacteria by a macrophage cytosolic surveillance pathway. Proc Natl Acad Sci U S A 2002; 99:13861–6.
Stockinger S, Materna T, Stoiber D, et al. Production of type I IFN sensitizes macrophages to cell death induced by Listeria monocytogenes. J Immunol 2002; 169:6522–9.
Auerbuch V, Brockstedt DG, Meyer-Morse N, ετ αλ. Mice lacking the type I interferon receptor are resistant to Listeria monocytogenes. J Exp Med 2004; 200:527–33.
Carrero JA, Calderon B, Unanue ER . Type I interferon sensitizes lymphocytes to apoptosis and reduces resistance to Listeria infection. J Exp Med 2004; 200:535–40.
McCaffrey RL, Fawcett P, O'Riordan M, et al. A specific gene expression program triggered by Gram-positive bacteria in the cytosol. Proc Natl Acad Sci U S A 2004; 101:11386–91.
O'Connell RM, Saha SK, Vaidya SA, et al. Type I interferon production enhances susceptibility to Listeria monocytogenes infection. J Exp Med 2004; 200:437–45.
O'Connell RM, Vaidya SA, Perry AK, et al. Immune activation of type I IFNs by Listeria monocytogenes occurs independently of TLR4, TLR2, and receptor interacting protein 2 but involves TANK-Binding Kinase 1. J Immunol 2005; 174:1602–7.
Stockinger S, Reutterer B, Schaljo B, et al. IFN regulatory factor 3-dependent induction of type I IFNs by intracellular bacteria is mediated by a TLR- and Nod2-independent mechanism. J Immunol 2004; 173:7416–25.
Zheng SJ, Jiang J, Shen H, Chen YH . Reduced apoptosis and ameliorated listeriosis in TRAIL-null mice. J Immunol 2004; 173:5652–8.
Remoli ME, Giacomini E, Lutfalla G, et al. Selective expression of type I IFN genes in human dendritic cells infected with Mycobacterium tuberculosis. J Immunol 2002; 169:366–74.
Weiden M, Tanaka N, Qiao Y, et al. Differentiation of monocytes to macrophages switches the Mycobacterium tuberculosis effect on HIV-1 replication from stimulation to inhibition: modulation of interferon response and CCAAT/enhancer binding protein β expression. J Immunol 2000; 165:2028–39.
Giacomini E, Iona E, Ferroni L, et al. Infection of human macrophages and dendritic cells with Mycobacterium tuberculosis induces a differential cytokine gene expression that modulates T cell response. J Immunol 2001; 166:7033–41.
Manca C, Tsenova L, Bergtold A, et al. Virulence of a Mycobacterium tuberculosis clinical isolate in mice is determined by failure to induce Th1 type immunity and is associated with induction of IFN-α /β. Proc Natl Acad Sci U S A 2001; 98:5752–7.
Bouchonnet F, Boechat N, Bonay M, Hance AJ . α/β interferon impairs the ability of human macrophages to control growth of Mycobacterium bovis BCG. Infect Immun 2002; 70:3020–5.
Stark GR, Kerr IM, Williams BR, et al. How cells respond to interferons. Annu Rev Biochem 1998; 67:227–64.
Player MR, Torrence PF . The 2-5A system: modulation of viral and cellular processes through acceleration of RNA degradation. May. Pharmacol Ther 1998; 78: 55–113.
Torrence PF, Maitra RK, Lesiak K, et al. Targeting RNA for degradation with a (2′-5′)oligoadenylate-antisense chimera. Proc Natl Acad Sci U S A 1993; 90:1300–4.
Kochs G, Haller O . Interferon-induced human MxA GTPase blocks nuclear import of Thogoto virus nucleocapsids. Proc Natl Acad Sci U S A 1999; 96:2082–6.
Yokoyama WM, Kim S, French AR . The dynamic life of natural killer cells. Annu Rev Immunol 2004; 22:405–29.
Zuniga EI, McGavern DB, Pruneda-Paz JL, et al. Bone marrow plasmacytoid dendritic cells can differentiate into myeloid dendritic cells upon virus infection. Nat Immunol 2004; 5: 1227–34.
Laing KJ, Secombes CJ . Chemokines. Dev Comp Immunol 2004; 28:443–60.
Servant MJ, Tenoever B, Lin R . Overlapping and distinct mechanisms regulating IRF-3 and IRF-7 function. J Interferon Cytokine Res 2002; 22:49–58.
Alcami A, Symons JA, Smith GL . The vaccinia virus soluble α/β interferon (IFN) receptor binds to the cell surface and protects cells from the antiviral effects of IFN. J Virol 2000; 74:11230–9.
Samuel CE . Antiviral actions of interferons. Clin Microbiol Rev 2001; 14:778–809.
Caraglia M, Marra M, Pelaia G, et al. α-interferon and its effects on signal transduction pathways. J Cell Physiol 2005; 202:323–35.
Sato M, Suemori H, Hata N, et al. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-α/β gene induction. Immunity 2000; 13:539–48.
Collins SE, Noyce RS, Mossman KL . Innate cellular response to virus particle entry requires IRF3 but not virus replication. J Virol 2004; 78:1706–17.
Acknowledgements
A. K. Perry is supported by the Howard Hughes Medical Institute predoctoral fellowship (Grant No. 59003787). Part of this work was also supported by National Institutes of Health research grants RO1 CA87924, RO1 AI056154, and R37 AI47868 to G. Cheng and from the Major Research Plan (30170461, 30430640) of Natural Science Foundation of China, and the National Basic Research Program of MOST (2002CB513001, 2001CB-510002) to H. Tang. H. Tang is also a fellow of Outstanding Young Investigators of National Naturual Science Foundation of China (30025010).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
PERRY, A., CHEN, G., ZHENG, D. et al. The host type I interferon response to viral and bacterial infections. Cell Res 15, 407–422 (2005). https://doi.org/10.1038/sj.cr.7290309
Issue Date:
DOI: https://doi.org/10.1038/sj.cr.7290309
Keywords
This article is cited by
-
A quantitative systems pharmacology model of the pathophysiology and treatment of COVID-19 predicts optimal timing of pharmacological interventions
npj Systems Biology and Applications (2023)
-
Mathematical analysis of within-host dengue dynamics with innate and adaptive immune responsess
The Journal of Analysis (2023)
-
Direct and indirect effects of IFN-α2b in malignancy treatment: not only an archer but also an arrow
Biomarker Research (2022)
-
The key role of Calpain in COVID-19 as a therapeutic strategy
Inflammopharmacology (2022)
-
A contemporary review on pathogenesis and immunity of COVID-19 infection
Molecular Biology Reports (2020)
