
ABSTRACT
The events of cell death and the expression of nuclear matrix protein (NMP) have been investigated in a promyelocytic leukemic cell line HL-60 induced with etoposide. By means of TUNEL assay, the nuclei displayed a characteristic morphology change, and the amount of apoptotic cells increased early and reached maximun about 39% after treatment with etoposide for 2 h. Nucleosomal DNA fragmentation was observed after treatment for 4 h. The morphological change of HL-60 cells, thus, occurred earlier than the appearance of DNA ladder. Total nuclear matrix proteins were analyzed by 2-dimensional gel electrophoresis. Differential expression of 59 nuclear matrix proteins was found in 4 h etoposide treated cells. Western blotting was then performed on three nuclear matrix acssociated proteins, PML, HSC70 and NuMA. The expression of the suppressor PML protein and heat shock protein HSC70 were significantly upregulated after etoposide treatment, while NuMA, a nuclear mitotic apparatus protein, was down regulated. These results demonstrate that significant biochemical alterations in nuclear matrix proteins take place during the apoptotic process.
Similar content being viewed by others

INTRODUCTIION
The nuclear matrix is an essential component of the nucleus which is important for the nuclear structural integrity and specific genomic functions1, 2. Several articles have reported that the nuclear matrix, as a higher order framework structures, might be disassembled during the apoptotic process3, 4, 5. Accordingly, nuclear lamins A/C or B have been found to decrease in apoptotic thymocytes6, T cells7, and carcinoma cell line 8, 9. The nucleolar protein B23, an obscure matrix protein has been reported to be degraded early in apoptotic prostate cells10. The above mentioned studies were mainly based on the use of immunocytochemical andultra- structural analysis11, 12. A detailed biochemical alterations of nuclear matrix protein (NMP) during apoptotic process have not been comprehensively investigated and therefore deserve further study.
High resolution two-dimentional gel electrophoresis (2-D gel) is an updated powerful tool for proteomics and functional analysis of the genome13, 14, since the technique had successfully been established to resolve 1100 different components from Escherichia coli15. Furthermore, in recent year, several proteins have been recognized as protein associated with nuclear matrix. Promyelocytic leukemia (PML) protein, a growth and transformation suppressor, was found in the nuclear bodies, which are mostly nuclear matrix-associated16. The PML and retinoic acid receptor-a (RARα) transcription factor leads gene are involved at the translocation breakpoint (15; 17) (q22; q12). This translocation event to the fusion of PML and retinoic acid receptor-α(RARα) genes and encoding of PML-RARa fusion protein17, 18. Nuclear matrix appears to be a target for heat shock effects and a determinant for stress response (HSC70, HSP70)19, 20. NuMA (nuclear mitotic apparatus protein), now known as NMP, was first described in 1980. It is in the nucleus and, during mitosis, it moves to the polar region of the mitotic spindle21.
In this report, we have used HL-60 cells as a model system to evaluate the dynamic alterations of nuclear matrix proteins after the induction of cell death with an antineoplastic agent etoposide. 2-D gel analysis of NMP was performed during the progress of apoptosis. The involvement ofsome nuclear matrix associated proteins was also studied by Western blotting analysis. The data thus far obtained indicated that the constituents of nuclear matrix proteins changed quite a lot during the early phases of apoptotic activation by etoposide. The disappearance of two unknown proteins and the change of the expression pattern of some nuclear matrix proteins are especially noticeable, which deserve further studies to understand the basic mechanisms leading to altered nucleus during apoptosis.
MATERIALS AND METHODS
Cell culture
HL-60 cells, a peripheral blood leukocyte from an acute promyelocytic leukaemia, were cultured in RPMI 1640 medium, 10% heat-inactivated fetal calf serum and 100 μg/ml streptomycin/penicillin, in 75 cm culture vessels in a humidified 5% CO2(v/v) incubator at 37°C. The cells were routinely kept in the logarithmic growth phase by diluting with fresh medium at least every third days.
Assessment of apoptosis by DNA fragmentation
Apoptosis was determined by genomic DNA fragmentation assessed by agarose gel electrophoresis. The sample in TE9S buffer containing 500 mM Tris-HCL, pH 9.0, 20 mM EDTA, 10 mM NaCl, 10% SDS and proteinase K (1 mg/ml) were incubated at 48 °C for 30 h, extracted twice with phenol/chloroform and once with chloroform described by Goelz et al22, treated for 2 h at 37°C with 250 mg/ml boiled bovine pancreatic RNase A , and loaded onto 1.5% (w/v) agraose horizontal slab gels containing 10 mg/ml ethidium bromide. DNA from 1×106 cell was loaded into each lane. Electrophoresis was performed in TAE buffer for 11 h at 15V. Gels were photographed under UV light.
Etoposide induction of apoptosis
Cells were harvested while in log phase (0.6–1.0×106 cell/ml), sedimented at 400×g and resuspended at a concentration of 1 ×106 cells/ml in complete medium containing 10 mM Hepes (pH 7.4). Etoposide (20 mg/ml), a semisynthetic podophllotoxin-derivative antineoplastic agent, was added in cell suspention (final concentration, 40 μg/ml). After varying length of incubation at 37°C, cells were sedimented at 3300× g for 3 min and washed with cold PBS.
TUNEL assay
In situ cell death detection, 1×106 cells which were induced apoptosis with 40 μg/ml of etoposide were harvested at 0, 0.5, 1, 2, 3 and 4 h respectively, The samples were washed 2 times with PBS and smeared on slide. Fix air dried cell sample with a freshly prepared paraformaldehyde solution (4% in PBS, pH 7.4) for 10 min at room temperature. Rinse slide twice with PBS. The samples were incubated with terminal deoxynucleotidyl transferase (TdT)-conjugated dUTP nick end labelling (TUNEL) with the in situ cell detection kit (Boehringer Mannheim Germany), processed as described in TUNEL method. The slide was subsequently counterstained with 1% methyl green. The apoptotic index (AI) was expressed as the percentage of positive nuclei for at least 500 counted cells.
Extraction of nuclear matrix protein
The method used was modified from Fey et al23. The cells induced with etoposide at 0, 3 and 4 h were collected and washed with PBS. The cells were lysed with cytoskeleton buffer (CSK) for 15 min at 4°C. The cell pellet was digested in digestion buffer for 20 min at 25°C. Chromatin-associated proteins were eluted by addition of ice-cold ammonium sulfate to the final concentration of 0.25 M. After spinned at 2200 rpm for 5 min at 4°C, the resulting pellet containing nuclear matrix and intermediate filament proteins was solubilized in disassembly buffer with 8 M urea, 1% β-mercaptoethanol (vol/vol) and 1 mM PMSF. The samples were then dialyzed against assembly buffer (1000:1 volume) for overnight at 4°C. By ultra-centrifugation at 207,000×g (SW41Ti rotor, BECKMAN) for 90 min, the supernatant containing nuclear matrix protein was removed carefully and concentration was determined by Dc protein assay Kit (Bio-Rad) with bovine serum albumin as a standard marker. The proteins were precipitated in 5 volumes of absolute ethanol, overnight at −20°C, and then centrifuged at 3500 rpm for 30 min. The pellet of NMPs was dried by N2 gas blowing. Dried protein was dissolved in first dimension sample buffer and was stored frozen at −80°C.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting
The samples of nuclear matrix protein were separated in minigels using 10% acrylamide (acrylamide/Bis 29:1, Bio-Rad ) on SDS-PAGE and electroblotted to nitrocellulose membrane, which were performed as described previously24. Protein sample of HL-60 cells were added respectively and carried out at 200Vhr. Electroblotting was performed with Mini Trans-blot Electrophoretic Transfer Cell (Bio-Rad). The membranes were blocked by 5% non-fat milk/TBST (0.05% Tween 20, 125 mM NCl, 25 mM Tris, pH 7.4), and were then incubated overnight at 4°C with HSC70 (B-6) mouse monoclonal IgG2a antibodies with 1:1500 dilution (Santa Cruz), NuMA (Ab-2) monoclonal mouse IgG with 1:600 dilution (Albiochem), or PML rabbit monoclonal antibody with 1:700 dilution (Santa Cruz). After washing 3 times, the membranes were then incubated with a 1:7500 dilution of sheep anti-mouse IgG-conjugated with horseradish peroxidase (Amersham Life Science) and goat anti-rabbit antibodies-conjugated with avidin-conjugated with horseradish peroxidase (BioGenex Lab., CA, USA) in TBST for 1 h respectively. The membrane was detected by Enhanced Chemiluminance (ECL) solution and exposed to high performance chemiluminescence film (Amersham) for 1 to 2 min. The visual reaction was scanned with an Imaging Densitometer GS-700 (Bio-Rad) and analysed. Western blotting of each antibody was performed three times.
Two-dimensional gel electrophoresis
High resolution 2-D gel electrophoresis carried out using Mini-Protean II Electrophoresis Cell (Bio-Rad). Non-equilibrium pH gradient gel electrophoresis (NEPHGE) was performed according to the method of O'Farrell15. Totally, NMP samples were extracted three times, and three gels were run for each sample. NEPHGE of 100mg sample was performed in glass tubes of 1.0 mm diameter at 1400 V/h, using ampholytes (0.4% pH 3–10 and 2% pH 5–7). The extruded gels were equilibrated 10 min at 20oC, and for the second-dimensional separation, then were placed on 10% polyacrylaminde slab gels (17 cm×8 cm). Silver staining was performed to visualise the resolved protein spots.
RESULTS
Analysis of apoptotic cells in HL-60 cells treated with etoposide
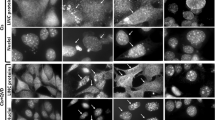
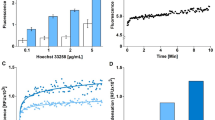
To derermine if the cell-death observed is associated with apoptosis, HL-60 cells treated ith etoposide was analyzed by the TUNEL assay. The increased amount of apoptotic cells with characteristic morphology was observed with increasing time (Fig 1). Apoptotic nuclei displayed a characteristic morphology comprising segregated and condensed chromatin masses localized adjacent to the nuclear envelope. Cell shrinkage, cytoplasmic compaction, and formation of apoptotic bodies were also found. Approximately 500 cells from each sample were analysed and the results are tabulated as percentage of apoptotic cells (Fig 2). TUNEL-positive cells (dark brown staining of DAB) at 0.5, 1, 2, 3 and 4 h after etopside treatments were about 6.85%, 18.54%, 38.89%, 34.15% and 33.00% respectively of the total cells. In the control sample (0 h), only a small number of apoptotic cells were found (less than 2.71%). The percentage of apoptotic cells at 2-4 h was not remarkably different and appeared to reach a plateau.

Morphologic indication of induced apoptosis. Monolayer of HL-60 cells (A-F) by (TdT) in situ cell death detection kit labeling of apoptosis induced DNA strand breaks (TUNEL), methyl green counter stain. Time after induction of apoptosis; A: 0 h, B: 0.5 h, C: 1 h, D: 2 h, E: 3 h, F: 4 h. Pictures show live(L), and apoptotic cells (A). Magnification, 1000×.

Time course of etoposide induced apoptotic index in HL-60 cells, an analysed by the TUNEL assay.
Analysis the fragmentation of genomic DNA by agarose gel electrophotesis
HL-60 cells were incubated with etoposide for varying length of time up to 22 h to induce apoptosis. Genomic DNA fragments extracted from etoposide treated HL-60 cells were analysed by agarose gel electrophoresos. As shown in Fig 3, After induced for 2 or 3 h by etoposide (Lane 3 and 4) the DNA from drug-treated cells was indistinguishable from the DNA of control cells (Lane 2). DNA ladder formation could be clearly observed when the HL-60 cells incubated for 4, 16, 20 and 22 h in cultures exposed to etoposide (lanes 5–8). The ladder of DNA fragments of approximately 200 bp multiples was observed.

Time course of etoposide induced DNA fragmentation in HL-60 cells in agarose gel electrophoresis. Lane 1: Molecular weight marker. Lane 2: DNA prepared from HL-60 cells untreated with etoposide. Lane 3, 4, 5, 6, 7 and 8 are DNAs from the cells incubated for 2, 3, 4, 16, 20 and 22 h with 40 mg/ml (68 μM) etoposide respectively.
Alteration of nuclear matrix proteins in etoposide treated HL-60 cells by SDS-PAGE
The nuclear matrix protein samples were separated by 12% SDS-PAGE slab gel and visualized by silver staining. The pattern of the nuclear matrix proteins revealed striking quantitative difference between control and apoptotic cells. Fig 4 showed nuclear matrix protein patterns from apoptotic cells with 4 h treatment (lane 3) in which a set of new bands of 32, 34, 35 KDa appeared. The bands of molecular weight of 28, 37, 40, 43, 50, 66, 70, 105, 116, 200, 210 KDa were more intense from apoptotic cells than control cells. In addition, the staining of bands of regions on 31, 60, 65, 95, 100 KDa also appeared to be increased as compared with control cells.

SDS-PAGE analysis of NMPs of HL-60 cells after apoptotic induction by etoposide. Lane 1: Molecular weight marker. Lane 2: DNA prepared from HL-60 cells untreated with etoposide. Lane 3, 4, 5, 6, 7 and 8 are DNAs from the cells incubated for 2, 3, 4, 16, 20 and 22 h with 40 mg/ml (68 mM) etoposide respectively.
Alteration of nuclear matrix proteins in etoposide treated HL-60 cells by 2-D gel electrophoresis
Nuclear matrix proteins were isolated and collected at 2 and 4 h after treatment with etoposide. The pattern of nuclear matrix proteins was significantly altered at 2 h and 4 h after treatment as compared with the uninduced control cells by 2-D gel electrophoresis. New spots appeared on the gel including 10 ∼ 24, 29, 46, 50 and 55 (shown in oval) at both 2 h and 4 h after treatment, as compared with control (See Fig 5A-C). Spots 1 ∼ 9, 27 and 30 only appeared at 2 h after treatment (compare Fig 5B with 5C). Another set of new protein spots appeared after incubation for 4 h, including spots 35 ∼ 38, 44, 45, 47, 56 and 57 (shown in rectangle in Fig 5C). The staining intensities of many spots such as 25, 26, 28, 31 ∼ 34, 39 ∼ 43, 48, 49 and 51 ∼ 54 were increased in 4 h treated samples, as compared with the pattern of the 2 h-treatment. However, two major proteins designated as spots A and B were found to be disappeared in etoposide treated cells (Fig 5,A-C). The co-ordinates of the altered nuclear-matrix proteins were shown in Tab 1.

2-D gel image nuclear matrix proteins of HL-60 cells before induction of apoptosis (control). Spots A and B appeared only in control. The positions of oval indicated that spots appeared at 2 and 4 h treatment. The positions of rectangle designated another set of new protein spots at 4 h treatment.

Nuclear matrix proteins of HL-60 cells 2 h after induction of apoptosis. Spots 1–9, 27 and 30 appeared only at 2 h as compared with control and 4 treatment. Spots 10–24, 29, 46, 50 and 55 present consistently both in 2 and 4 h treated sample, as compared with control. The positions of rectangle indicated that spots were appeared after induction of apoptosis 4 h. Spots A and B (arrows indicated) disappeared in etoposide treated cells.

Nuclear matrix proteins pattern 4 h after induction of apoptosis New spots appeared on position of spots 10–24, 29, 46, 50 and 55 (shown in oval), spots 1–9, 27 and 30 disappeared at 4 h treatment. Another set of new protein spots appeared after incubation for 4 h which was designated as spots 35-38, 44, 45, 47, 56 and 57 (shown in rectangle). However, two major proteins designated spots A and B were found to be disappeared in etoposide treated cells.
Altered expression of PML, HSC 70 and NuMA in nuclear matrix of etoposide treated HL-60 cell
To further investigate if there are additional alterations of nuclear NMPs during etoposide treatment of HL-60 cells, Western blotting of three different nuclear matrix associated proteins were analyzed including PML, NuMA and HSC70 proteins. The expression of the 110 KDa PML protein and 70 KDa HSC70 protein increased dramatically in HL-60 cells after etoposide induction (Fig 6A and B). However, a significant reduction of the 238 KDa NuMA proteins was found in the nuclear matrix of HL-60 cells under the same treatment (Fig 6C).

Western blotting analysis for PML (rabbit monoclonal antibody with 1:700 dilution, (A)HSC 70 B-6 mouse monoclonal IgG2a antibodies with 1:1500 dilution, (B)and NuMA (Ab-2 monoclononal mouse IgG with 1:600 dilution, (C) in normal nuclear matrix protein (lane 1) from untreated HL-60 cells, and apoptotic unclear matrix protein (lane 2) from etoposide induced 4 h HL-60 cells.
DISCUSSION
We have demonstrated the potential alterations of some nuclear matrix proteins that could be detected from apoptotic samples by high resolution 2-D electrophoresis. 48 new nuclear matrix protein spots appeared after etoposide treatment for 2 h, as compared with control. Again, another 9 new spots were detected after 4 h of induction of apoptosis, and at the same time, 11 spots that once appeared at 2 h were disappeared. In addition, the intensities of some pre-existing spots increased. At 2 h and 4 h the spots A and B were no longer detectable. Differential expression of 59 nuclear matrix proteins was found in etoposide treated cells for 4 h, as compared with untreated HL-60. The alteration of protein composition may be directly associated with apoptosis, although we could not rule out other possibilities such as DNA repair, cell-cycle arrest.Spots newly appeared during the progress of
apoptosis may represent proteins involved in the process of active cell death such as the caspases or proteases and their cleaved products. Components of the RNA processing system might also associate with the nuclear matrix 23, 25, 26. Nevertheless, it is possible that the majority of NMP spots which were present consistently on three 2-D gel patterns (0, 2, 4 h after etoposide treatment) may not be related to apoptosis. The relationship between the DNA fragmentation by in situ DNA-end labeling and DNA ladder on agarose gels has been reported for apoptotic bone marrow cells and erythroid cells27, 28. Our results are in agreement with previous findings, that the characteristic morphology change appeared earlier than DNA fragmentation. Following the time course of apoptosis, nuclei display a characteristic morphological changes at 2 h after induction, but without DNA ladder on agarose gels. Four hours after apoptotic activation, DNA is being cleaved into oligonucleosomal fragments and apoptotic bodies formed, then the amount of apoptotic cells reaches a plateau (33%), and NA ladder can be observed. Therefore, it is worthy to note that the alteration of the nuclear matrix proteins composition occurs earlier (2 h) than the DNA fragmentation (4 h) during apoptosis of HL-60 cells.
We have performed SDS-PAGE analysis on control and apoptotic samples of nuclear matrix protein. It is evident that the silver staining bands display a high degree of differences. It is worth emphasizing that the intens bands of 70, 110 KDa show striking increase from apoptotic sample at 4h, a result which can be seen from Western blotting analysis as shown in Fig 6, A and B as detected by PML and HSC70 antibodies. In this figure, nuclear mitotic apparatus (NuMA) protein is found to be down-regulated from nuclear matrix protein of HL-60 cells exposed to etoposide for 4 h. On the other hand, the expression level of the HSC70 in the nuclear matrix proteins is upregulated in etoposide treated cells as compared to the control, suggesting the detrimental effects of etoposide and the increased of HSC70 may serve to protect against such stress effect. The recent report by Kun-Sang Chang et al16 showed that PML is associated with the nuclear matrix protein of the NIH/3T3 cells transfected with pSG5PML expression plasmids by Western-blotting analysis. Suurprisingly, α significant increase of 110 KDa PML-RAR a fusion protein in the nuclear matrix of the untransfected HL-60 cells treated with etoposide has been also found in the present work. Thisnuclear matrix protein may be either associated with G1 cell-cycle arrest as mentioned recently29, or with the increase of apoptotic cells since it has been shown that PML protein facilitates programmed cell death30.
In conclusion, the data in this experiment indicate that during etoposide induction of apoptosis in HL-60 cells, significant alterations in the expression of nuclear matrix proteins have been found. The results not only revealed the changing patterns of nuclear matrix proteins during various time course of chromatin condensation and DNA fragmentation after induction of cell death in HL-60 cells, but also identified the alteration of some functionally important molecules, including the structural element NuMA, the suppressor protein PML and molecular chaperone HSC70 during the apoptotic process.
References
Stuurman N, De Jong L and Van Driel R . Nuclear frameworks: concepts and operational definitions. Cell Biol Int Rep 1992; 16:837–52.
Berezney R and Jeon KW .Structural and functional organization of the nuclear matrix. International Review of Cytology. Academic Press, San Diego, CA 1995; 162A and 162B, 1–65.
Miller TE, Beausang LA, Meneghini M, Lidgard G . Death-induced changes to the nuclear matrix: the use of anti-nucleer matrix antibodies to study agents of apoptosis. Biotechniques 1993; 15:1042–7.
Tinnemans MMFJ, Lenders M-HJH et al. Alteration in cytoskeletal and nuclear matrix- associated proteins during apoptosis. EUR J Cell Biol 1993; 68:35–46.
Weaver VM, Carson CE, Walker PR et al. Degradation of nucler matrix and DNA cleavage in apoptotic thymocytes. J Cell Sci 1996; 109:45–56.
Neamati N, Fernandez A, Wright S et al. Degradation of lamin B1 precedes oligonucleosomal DNA fragmentation in apoptic thymocytes and isolated thymocyte nuclei. J Immunol 1995; 154:3788–95.
Casiano C, Marti S, Green D, et al. Selective cleavage of nuclear autoantigens during CD95 (Fas/APO-1)-mediated T cell apoptosis. J Exp Med 1996; 184:765–70.
Tinnemans M, Lenders J, Ten Veld G et al. Alteration in cytoskeletal and nuclear matrix associated proteins during apoptosis. J Cell Biol 1994; 126:827–37.
Lazebnik Y A, Takahashi A, Moir R et al. Studies of the lamin proteinase reveal multiple parallel biochemical pathways during apoptotic execution. Proc Natl Acad Sci USA 1995; 92:9042–6.
Tawfic S, Olson M, and Ahmed K . Role of protein phosphorylation in post- translational regulation of ptotein B23 during programmed cell death in the prostate gland. J Biol Chem 1995; 270:21009–15.
Earnshaw WC : Nuclear changes in apoptosis. Curr Opin Cell Biol 1995; 7:337–43.
Falcieri E, Zamai L, Santi S, et al. The behaviour of nuclear domains in the course of apoptosis. Histochemistry 1994; 104:221–31.
Anderson NI, Anderson NG . Proteome and proteomics: new technologies, new concepts, and new worlds. Electrophoresis 1998; 19:1853–61.
Klose J, Kobalz U . Two-dimensional electrophoresis of proteins: an updated protocal and implications for a functional analysis of the genome. Electrophoresis 1995; 16:1034–59.
O'Farrell PH . High resolution two-dimensional electrophoresis of proteins. J Biol Chem 1975; 250(10):4007–21.
Kun-Sang Chang, You-Hong Fan, Michael Andreeff et al. PML gene encodes a phosphoprotein associated with the nuclear matrix. Blood 1995; 85(12):3646–53.
Borrow J, Goddard AD, Sheer D et al. Molecular analysis of acute promyelocytic leukemia breakpoint cluster region on chromosome 17. Science 1990; 249(4976):1577–80.
De th H, Chomienne C, Lanotte M et al. The t(15;17) translocation of acute promyelocytic leukaemia fuses the retinoic acid receptor alpha gene to a novel transcribed locus. Nature 1990; 347(6293):558–61.
Yano M, Naito Z, Tanaka S et al. Expression and roles of heat shock proteins in human breast cancer. Jpn J Cancer Res 1996; 87(9):908–15.
Roti JL, Wright WD, VanderWal R et al. The nuclear matrix: a target for heat shock effects and a determinant for stress response. Crit Rev Eukaryot Gene Expr 1997; 7(4):343–60.
He D, Zeng C and Brinkley BR . Nuclear matrix proteins as structural and functional components of the mitotic apparatus. Int Rev Cytol 1995; 102B:1–73.
Goelz SE, Hamilton SR, Vogelstein B . Purification of DNA from formaldehyde fixed and paraffin embedded human tissue. Biochem Biophys Res Commun 1985; 130:118–26.
Fey EG, Penman S . Nuclear matrix proteins reflect cell type of origin in cultured human cells. Proc Natl Acad Sci USA 1988; 85:121–5.
Harlow david Lane : Antibodes. A laboratory Manual. 1988; 479–492, Cold spring Harbor Laboratory Press, USA.
Kiyokazu Morioka, Shigenobu Tone, Masahiro Mukaida et al. The apoptotic and nonapoptotic nature of the terminal differentiation of Erythroic cells. Exp Cell Res 1998; 240:206–17.
Stein GS, Stein JL, Lian JB et al. Functional interrelationships between nuclear structural and transcriptional control: Contributions to regulation of cell cycle and tissue specific gene expression. J Cell Biochem 1997; 62:198–209.
Lepelley P, Campergue L, Grardel N et al. Is apoptosis a massive process in myelodysplastic syndrome Brit. J Haematol 1996; 95:368–71.
Nickerson JA, Krockmalnic G, Wan KM . Penman S . The nuclear matrix revealed by eluting chromatin from cross-linked nucleus. Proc Natl Acad Sci USA 1997; 94:4446–50.
John Y H Chan, Li Li, Yu-Hung Fan et al. Cell-cycle regulation of DNA damage-induced expression of the suppressor gene PML. Biochem and Biophy Res commun 1997; 240:640–46.
Andre C, Guillemin MC, Zhu J et al. The PML and PML/RARalpha domains: from autoimmunity to molecular oncology and from retinoic acid to arsenic. Exp Cell Res 1996; 229(2):253–60.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
JIN, M., ZHANG, P., DING, M. et al. Altered expression of nuclear matrix proteins in etoposide induced apoptosis in HL-60 cells. Cell Res 11, 125–134 (2001). https://doi.org/10.1038/sj.cr.7290077
Received:
Revised:
Accepted:
Issue Date:
DOI: https://doi.org/10.1038/sj.cr.7290077
