
Abstract
At present, nuclear condensation and fragmentation have been estimated also using Hoechst probes in fluorescence microscopy and flow cytometry. However, none of the methods used the Hoechst probes for quantitative spectrofluorometric assessment. Therefore, the aim of the present study was to develop a spectrofluorometric assay for detection of nuclear condensation and fragmentation in the intact cells. We used human hepatoma HepG2 and renal HK-2 cells cultured in 96-well plates treated with potent apoptotic inducers (i.e. cisplatin, staurosporine, camptothecin) for 6–48 h. Afterwards, the cells were incubated with Hoechst 33258 (2 µg/mL) and the increase of fluorescence after binding of the dye to DNA was measured. The developed spectrofluorometric assay was capable to detect nuclear changes caused by all tested apoptotic inducers. Then, we compared the outcomes of the spectrofluorometric assay with other methods detecting cell impairment and apoptosis (i.e. WST-1 and glutathione tests, TUNEL, DNA ladder, caspase activity, PARP-1 and JNKs expressions). We found that our developed spectrofluorometric assay provided results of the same sensitivity as the TUNEL assay but with the advantages of being fast processing, low-cost and a high throughput. Because nuclear condensation and fragmentation can be typical markers of cell death, especially in apoptosis, we suppose that the spectrofluorometric assay could become a routinely used method for characterizing cell death processes.
Similar content being viewed by others

Introduction
Apoptosis is a complex process including various morphological and biochemical cellular changes that can be used for apoptosis characterization. The morphological changes include cell shrinkage, pyknosis and karyorrhexis followed by DNA fragmentation in late stage of apoptosis1,2. Then, the cellular cytoskeleton is damaged leading to membrane blebbing and, finally, apoptotic bodies formation1,2,3.
Pyknosis is an irreversible process of nuclear condensation commonly associated with early stages of apoptosis and necrosis4. Pyknosis can be divided into nucleolytic and anucleolytic pyknosis5. Nucleolytic pyknosis mainly occurs during apoptosis. Chromatin is condensed into large clumps which can be packed into apoptotic bodies. In contrast, anucleolytic pyknosis predominantly occurs during necrosis when chromatin condenses into smaller, irregular clumps4,6.
Karyorrhexis occurs during late stage of apoptotic and necrotic processes when nucleus is fragmented and chromatin irregularly distributed into the cytoplasm, followed by formation of apoptotic bodies and karyolysis, respectively. Karyolysis is a complete enzymatic degradation of chromatin in dying cells7. Nuclear condensation and fragmentation have been used as markers for late stages of apoptosis8.
Caspases play crucial roles in apoptosis. Caspases 8, 9 and 10 initiate apoptotic cascade through activation of effector caspases 3, 6 and 79. DNA fragmentation factor (DFF) has an essential role in DNA cleavage during the apoptotic process. Activated caspase 3 cleaves the DNA fragmentation factor, and activates its catalytic subunit (DFF40)10. Activated DFF40 cleaves double-stranded DNA with a preference for adenine and thymine (A/T) rich region in internucleosomal linker regions into approx. 180 bp fragments and multiples thereof1,11. This characteristic DNA cleavage pattern is called “DNA ladder” and can be used for detection of apoptotic DNA fragmentation1,12,13. In addition to DNA ladder, comet and Terminal deoxynUcleotidyl transferase Nick-End Labeling (TUNEL) assays belong among frequently used methods for apoptotic DNA fragmentation detection. DNA ladder assay uses the presence of the DNA ladder fragments pattern occurring during apoptosis detected after electrophoresis in agarose gel, while comet detects fragmented DNA in the gel. In the TUNEL technique, free -OH moiety in the double and single strand DNA breaks is labeled using modified deoxynucleotides analogues tagged with various markers allowing DNA strand breaks detection14. Chromatin condensation and DNA fragmentation can be also characterized using microscopic methods, especially fluorescence microscopy. Hoechst fluorescent probes originate from bisbenzimides, a family of lipophilic substances, that bind preferentially to a small groove of A/T rich DNA sequences called A-T regions15. At least three consecutive A-T base pairs are required for specific Hoechst dye binding leading to fluorescence increase. In addition, some papers reported that the nucleus of apoptotic cells can exhibit enhanced fluorescence after Hoechst binding16,17,18. Therefore, Hoechst probes have been frequently used for nucleus staining in fluorescence microscopy and flow cytometry19,20,21. At present, three Hoechst probes have been used in vitro: 3325822, 3334223, 3458024. Hoechst 33258 is a lipophilic and cell permeable probe25,26,27 most frequently used in fluorescence microscopy for qualitative detection of nuclear morphology changes, primarily for detecting cell shrinkage, chromatin condensation, nuclear fragmentation and apoptotic bodies formation in various cell lines28,29,30,31,32. Despite the unique Hoechst probe properties for nuclear changes detection, no scientific study, however, reported a quantitative spectrofluorometric method development. Thus, the aim of the present study was to develop a Hoechst 33258 dye spectrofluorometric assay for quantitative measurement of nuclear condensation and fragmentation in intact cells. Finally, we aimed to evaluate the obtained outcomes in comparison with other frequently used methods for cell damage (i.e. WST-1 and glutathione assays) and apoptosis (TUNEL, DNA ladder, etc.) detection.
Results
Optimization of the spectrofluorometric assay
We aimed to develop a spectrofluorometric method for detection of nuclear condensation and fragmentation in the intact cells using a fluorescent probe Hoechst 33258. According to the literature, we used the same λ(ex,max) = 352 nm and λ(em,max) = 461 nm in H33258 measurements33,34,35, but optimized the experimental procedure, i.e. H33258 concentration and incubation length of the probe with cells. To induce cell damage, we incubated HepG2 cells with 100 µM cisplatin (CisPt) for 24 h. Then, we incubated the cells with the concentrations range of 0.1–5 µg/mL H33258 (Fig. 1A). The highest fluorescence signal was detected in 5 µg/mL H33258 in CisPt treated cells but the background fluorescence in untreated HepG2 cells was strongly enhanced too. Due to the highest signal-to-noise ratio found after the 2 µg/mL, we selected this H33258 treatment concentration to be optimal for following experiments.

Optimization of the spectrofluorometric assay for detection of nuclear condensation and fragmentation in HepG2 cells. (A) Optimization of H33258 concentration (0.1; 1; 2; 5 µg/mL) – untreated cells (white columns), 100 µM cisplatin treated cells (blue columns). Mean ± SEM (n = 3). (B) Change of intensity of fluorescence over time. HepG2 cells were treated with CisPt for 24 h. Then, the cells were incubated with H33258 (2 µg/mL) and fluorescence (EX/EM = 352/461 nm) was recorded for 10 min. Untreated cells (black), 50 µM CisPt (light blue), 100 µM CisPt (dark blue). (C,D) Detection of nuclear condensation and fragmentation in cells. HepG2 cells were treated with cisplatin for 24 h. Then, the cells were incubated with H33258 (2 µg/mL) and fluorescence (EX/EM = 352/461 nm) was recorded for 5 min. Finally, the background fluorescence was subtracted and fluorescence intensity corresponding to the extent of nuclear condensation and fragmentation was showed in untreated (white), 50 µM (light blue) and 100 µM CisPt (dark blue) treated cells (= C). The fluorescence intensity attributed to the extent of nuclear condensation and fragmentation was compared among untreated cells (white), 50 µM CisPt (light blue), 100 µM CisPt (dark blue) treated cells (= D).
For estimation of optimal cell incubation time with H33258, HepG2 cells were treated with CisPt (0; 50; 100 µM) for 24 h. We found that centrifugation of cells (5 min, 8000g; RT) after the treatment with tested compounds, followed by cell culture medium replacement, is crucial for achieving repeatable results because it ensures the sedimentation of all cells on the bottom of a well. After centrifugation, 70 µL of culture medium was replaced with 70 µL of PBS 1 × in each well. Then, 10 µL of H33258 was added to obtain final concentration 2 µg/mL H33258 in a well and fluorescence was recorded at EX/EM = 352/461 nm.
We found that fluorescence intensity was increasing strongly during the first minute of cells incubation with H33258 (Fig. 1B). Then, the fluorescence intensity (IF) remained rather stable between the second and the tenth minute of incubation implying that any of durations including this time interval could be used for the purposes of the spectrofluorometric assay. We selected 5 min of H33258 incubation with cells in all following experiments. In addition, we confirmed that IF was enhanced relatively to the increasing CisPt concentration in cells according to expected induction of nuclear condensation and fragmentation. Figure 1C shows IF detected in cells after subtraction of background fluorescence intensity in blank samples (i.e. without cells). Finally, the extent of nuclear condensation and fragmentation in cells was expressed in Relative Fluorescence Units (RFU) (Fig. 1D).
Estimating sensitivity of the spectrofluorometric assay
Further aim of our study was to use the H33258 spectrofluorometric assay in cells exhibiting nuclear condensation and fragmentation of different origin and extent. We incubated HepG2 and HK-2 cells with CisPt (0; 0.5; 5; 25 and 100 µM) for 24 and 48 h. After H33258 treatment, we observed increasing IF relating to both CisPt doses and incubation time in HepG2 (Fig. 2A) and HK-2 cells (Fig. 2B). We found a significant increase of IF in 25 and 100 µM CisPt treated cells after 24 and 48 h. In addition, the extent of condensation and fragmentation detected after 24 and 48 h demonstrated time and dose dependent increase (Fig. 2A,B).

Characterizing cisplatin-induced toxicity in HepG2 and HK-2 cells. HepG2 (A,C,E) and HK-2 (B,D,F) cells were incubated with cisplatin (CisPt; 0; 0.5; 5; 25 and 100 µM) for 24 and 48 h. After treatment, nuclear condensation and fragmentation using the H33258 spectrofluorometric assay (A,B), dehydrogenase activity using the WST-1 test (C,D) and glutathione levels using monochlorobimane assay (E,F) were measured. The data are presented as mean ± SEM (A, B) and mean ± SD (C-F). (*p < 0.05; **p < 0.01; ***p < 0.001, vs. untreated cells at appropriate time interval).
We used two additional biochemical assays to characterize CisPt toxicity in cells, i.e. the WST-1 test detecting intracellular dehydrogenase activity and glutathione assay. We found considerably reduced dehydrogenase activity in HepG2 treated with ≥ 25 µM CisPt (Fig. 2C) and for all CisPt treatment of HK-2 cells in 24 h treatment (Fig. 2D), while for both cells type in 48 h treatment, the doses ≥ 5 µM CisPt had significant reduction. On the other hand, the HK-2 cell line exhibited lower susceptibility to glutathione depletion than HepG2 cells because a significant reduction of glutathione levels was found only at 100 µM CisPt (Fig. 2E,F) for 24 h treatment, while for HepG2 the significant reduction was ≥ 25 µM. For 48 h treatment, both cell types had significant reduction in similar CisPt concentrations treatments ≥ 5 µM. In conclusion, the outcomes found using the WST-1 and glutathione assays confirmed the occurrence of CisPt toxicity detected using the H33258 spectrofluorometric assay. WST-1 and glutathione assays detected cellular damage also in cells treated with 5 µM CisPt. Hence, both biochemical assays are more sensitive in detection of a cell damage in comparison to H33258 assay detecting structural nuclear changes.
In addition to CisPt, we aimed to utilize the spectrofluorometric assay for detection of nuclear changes in cells treated with other apoptotic inducers. Thus, we incubated HepG2 and HK-2 cells with camptothecin (CAM, 1; 5 µM) and staurosporine (STA, 10; 100 nM) for 6, 24 and 48 h (Fig. 3A,B). We used CisPt as a positive control and 10 µg/mL and TiO2 P25 nanoparticles as a negative control. All results were compared to the signal in untreated cells at appropriate time interval. Our results showed that we detected a significant increase of nuclear condensation and fragmentation in all tested compounds except of TiO2 P25 nanoparticles which did not induce any detected nuclear changes at all tested time durations. After 6 h of treatment, we found a significant increase of IF related to enhanced nuclear changes only in HK-2 cells treated with 5 µM CAM (p < 0.001). At 24 h, a significant increase of nuclear condensation and fragmentation comparing with untreated cells was detected in all HK-2 treated cells with exception of 1 µM CAM (p = 0.198) and 10 nM STA (p = 0.999). We also found a significant increase of IF in HepG2 cells treated with higher concentrations of used apoptotic inducers. Thus, the H33258 assay did not detect a significant nuclear condensation and fragmentation in 1 µM CAM (p = 0.935), 10 nM STA (p = 0.135) and 50 µM CisPt (p = 0.382) treated HepG2 cells for 24 h.

Detection of nuclear condensation and fragmentation using the H33258 spectrofluorometric assay. HepG2 (A) and HK-2 (B) cells were treated with cisplatin (CisPt, 50; 100 µM, blue columns), camptothecin (CAM, 1; 5 µM, green columns), staurosporine (STA, 10; 100 nM, yellow columns) for 6, 24 and 48 h. Control (CTRL) represents untreated cells. The cells treated with TiO2 P25 nanoparticles (NPs, 10 µg/mL, orange columns) were used as a negative control. After treatment, the cells were incubated with H33258 (2 µg/mL) for 5 min and intensity fluorescence (EX/EM = 352/461 nm) was measured. Then, the background fluorescence was subtracted and the extent of nuclear condensation was expressed in RFU. Data are presented as mean ± SEM (n = 8–20). (*p < 0.05; **p < 0.01; ***p < 0.001; vs. untreated cells at appropriate time interval).
After 48 h of treatment, we found significantly increased extent of nuclear condensation in almost all tested concentrations of compounds in both cell lines. The only exception was observed in 10 nM STA treated HepG2 cells (p = 0.999) which exhibited a decrease of IF in comparison with 10 nM STA treated cells for 24 h. All tested concentrations of pro-apoptotic compounds induced larger nuclear condensation after 48 h incubation in comparison to 24 h and the cells treated with higher concentrations of tested compounds exhibited larger nuclear impairment in comparison to lower dose. The highest IF signal was detected in HepG2 and HK-2 cells treated with 100 µM CisPt and 5 µM CAM at 48 h. In conclusion, our results confirmed that the H33258 spectrofluorometric assay was capable to detect increased nuclear condensation and fragmentation in all tested apoptotic inducers relatively to concentration and incubation period (Fig. 3A,B).
Comparison of H33258 spectrofluorometric assay with apoptosis detecting methods
To evaluate the outcomes of newly developed H33258 spectrofluorometric assay, we aimed to compare the results with other methods characterizing pro-apoptotic processes in cells. Therefore, we treated HepG2 and HK-2 cells with CisPt (50 and 100 µM) for 6, 24 and 48 h and characterized nuclear changes and pro-apoptotic activation using four additional measurements, i.e. caspase 3/7 activity, PARP-1 and JNK proteins expression, TUNEL assay and DNA ladder.
We measured the activity of caspases 3 and 7 to evaluate apoptotic activation in the cells. Despite detecting a significant increase in caspase 3/7 activity in both cell lines treated with 100 µM CisPt for 6 h, the largest enhancement of caspase 3/7 activity was detected in cells treated with 50 µM CisPt for 24 h (Fig. 4). At 48 h, the caspase 3/7 activities remained increased although we found a decrease in comparison to cells treated for 24 h.

Caspase 3/7 activity in CisPt treated cells. HepG2 (A) and HK-2 (B) cells were treated with CisPt (50 and 100 µM) for 6, 24 and 48 h. After incubation, the activity of caspases 3/7 was measured (EX/EM = 485/535 nm). Data are presented as mean ± SEM (n = 2). (*p < 0.05; **p < 0.01; ***p < 0.001; vs. untreated cells at appropriate time interval).
Then, we analyzed the levels of PARP-1, PARP-1 fragment (fPARP-1), pJNK and β-actin. PARP-1 protein serves as a substrate for caspase 3 during apoptosis. Thus, PARP-1 cleavage leading to fPARP-1 production is related to the extent of caspase 3 activation. After 24 h of treatment, we detected PARP-1 fragmentation in cells treated with both concentrations of cisplatin which persisted until 48 h only in HepG2 cells (Table 1). Phosphorylation of JNK1 and JNK2 can also correspond with activated apoptotic process and that is why we aimed to estimate their levels. We observed that pJNK1 levels were increased in all tested time incubation with CisPt in HepG2 cells. In HK-2 cells, protein expressions of pJNK1 and pJNK2 were stimulated predominantly at 24 h of treatment. Thus, the detected increase of pJNK levels correlated strongly with PARP fragmentation in both cell lines.
TUNEL assay was used for detection of DNA strand breaks in CisPt treated cells (Fig. 5). After 6 h of treatment, a weak fluorescence demonstrating presence of DNA strand breaks was observed only in 100 µM CisPt treated HepG2 cells. At longer time durations, TUNEL staining detected strong DNA fragmentation in both cell lines treated with both 50 and 100 µM CisPt (Fig. 5).

TUNEL assay in CisPt treated cells. HepG2 and HK-2 cells were treated with CisPt (50 and 100 µM) for 6, 24 and 48 h. Then, the cells and nuclei were visualized using phase contrast (left, magnification 400 ×), TUNEL (FITC 480/30 nm; middle) and Hoechst 33258 stainings (DAPI 375/28 nm; right). Scales correspond to 25 µm.
At final, we used DNA ladder assay in CisPt treated cells to estimate the extent of DNA fragmentation. The internucleosomal DNA fragmentation can occur as the terminal feature of apoptosis. Thus, we detected DNA ladder pattern only in CisPt treated HepG2 cells after 48 h (Fig. 6A) and in HK-2 cells after 24 and 48 h (Fig. 6B).

DNA ladder assay in CisPt treated cells. HepG2 (A) and HK-2 (B) cells were treated with CisPt (50 and 100 µM) for 6, 24 and 48 h. After DNA isolation, DNA ladder assay was performed. (0, untreated cells; 50, 50 µM CisPt; 100, 100 µM CisPt; M, 100 bp marker). Original gels images were included into the Supplementary file.
We summarized and compared the results obtained using all methods detecting apoptosis in CisPt treated HepG2 and HK-2 cells (Table 2). We proved that the initial increase in caspase 3/7 activity was followed by fragmentation of PARP-1, DNA fragmentation detected using TUNEL assay and formation of DNA ladder. In addition, Table 2 shows that the newly developed spectrofluorometric assay using the H33258 dye provided outcomes fully comparable with TUNEL assay which has been used as a standard method detecting increased levels of DNA fragmentation in cells.
Discussion
DNA staining using Hoechst fluorescent probes belongs to commonly used approaches for visual detection of nuclear condensation. Recent scientific studies have described the use of H33258 for fluorescence detection of cell shrinkage, chromatin condensation, nuclear fragmentation and apoptotic bodies formation in cells16,28,29,30,31,32. Surprisingly, these reports used H33258 only for microscopic detection of nuclear changes. Thus, we aimed to develop a quantitative spectrofluorometric method for the detection of nuclear condensation and fragmentation using H33258 in cultured cells.
In this study, we used HepG2 and HK-2 cells. Doubling times of HepG2 and HK-2 cells are ca 48 h and 54 h, respectively36,37. Thus, the influence of cell cycle duration on obtained results was rather negligible. Firstly, we optimized H33258 concentration which is of crucial importance due to potential toxicity of the dye. Some studies reported that H33258 levels higher than 5 µg/mL are toxic to cells especially after long-term exposure38,39. H33258 concentration used in studies has been 1–5 µg/mL H33258 in fluorescence microscopy40,41 and 0.5–2 µg/mL H33258 in flow cytometry21,42,43. Therefore, we decided to estimate H33258 concentrations in the range of 0.1–5 µg/mL for development of the spectrofluorometric assay. We selected 2 µg/mL H33258 to be optimal for treatment of cells without risk of cell impairment during the spectrofluorometric measurement for 5 min.
After introduction of the spectrofluorometric assay´s conditions, we aimed to utilize the assay for detection of chromatin condensation and nuclear fragmentation in two cell lines of different origin, i.e. human liver carcinoma HepG2 cells and proximal tubular epithelial HK-2 cells, treated with three inducers of apoptosis (cisplatin, camptothecin, staurosporine). Cisplatin is an anticancer drug capable to induce apoptotic DNA fragmentation in cells44,45. CisPt interracts with adenine or guanine forming adducts which inhibit DNA replication and transcription leading to p53 or MAPK activation46. Camptothecin is an inhibitor of DNA topoisomerase I47,48,49 causing formation of DNA strand breaks and p53 activation. Staurosporine is a strong inhibitor of protein kinases inducing DNA fragmentation50,51. Moreover, the cells were incubated with TiO2 P25 nanoparticles which have been reported to possess no capability to induce nuclear condensation or DNA fragmentation in HepG2 cells52.
According to our results, we showed that the developed spectrofluorometric assay using H33258 was capable to detect nuclear changes. In addition, the intensity of fluorescence reflected different extent of nuclear condensation and fragmentation in comparison of 6, 24 and 48 h of incubation in CAM, STA and CisPt treated cells. Our finding of nuclear condensation and fragmentation detected using the new spectrofluorometric method after treatment with 5 µM CAM for 6 h can be supported by a study of Rath et al.48 who found presence of apoptotic nuclei in 5 µM CAM treated thyroid carcinoma cells after 8 h of treatment using TUNEL assay. Another study focused on estimation of CAM-dependent induction of apoptosis showed occurrence of apoptotic changes in 1 µM CAM treated HepG2 cells after 24 h53 using flow cytometry. Therefore, the newly developed H33258 spectrofluorometric method provided similar outcomes to other scientific reports studying apoptosis induced by camptothecin.
To characterize staurosporine effect in cells, we used the concentrations which have been reported to be sufficient to induce the cell death50,54. Our results on detection of changes in nuclear structure using the spectrofluorometric method are consistent with a study of Ding et al.50 who also detected DNA fragmentation in 10 nM STA treated HepG2 cells after 24 h of treatment using TUNEL assay. Another paper of Deshmuk and Johnson51 described a finding of increased DNA fragmentation in 100 nM STA treated sympathetic neurons after 24 h using TUNEL staining as well. Therefore, we can conclude again that the spectrofluorometric method using H33258 provided results similar to the outcomes of other studies characterizing staurosporine action in cells.
Finally, we aimed to use the spectrofluorometric assay using the H33258 dye for characterization of cisplatin action in cells and to compare obtained results with the outcomes of other apoptosis detecting assays. Occurrence of significant pro-apoptotic nuclear changes in cisplatin-treated cells has been broadly reported after 24 h of incubation. A study of Yang et al.45 detected increased DNA fragmentation in 50 µM CisPt treated HK-2 cells for 24 h using TUNEL assay. Similar results were obtained also in CisPt treated LLC-PK1 kidney cells using TUNEL assay55 and in HepG2 cells using fluorescence microscopy and flow cytometry44. Based on these papers, our findings of increased nuclear condensation and fragmentation in 50 and 100 µM CisPt treated HepG2 and HK-2 cells using the developed spectrofluorometric assay are fully consistent with the literature.
The latter goal of our study was to compare the outcomes obtained using the developed spectrofluorometric assay in CisPt treated cells with outcomes of other, frequently used assays detecting apoptosis, i.e. caspase 3/7 activity, JNK/PARP proteins expression, TUNEL assay and DNA ladder. Caspase 3/7 activation detected in our experiments after 6 h of treament was also described in a paper of Schweyer et al.56 in 50 µM CisPt treated NCCIT cells. Persisting activation of caspases 3/7 was reported in a number of studies, e.g. in UBOC1, HK-2 and SH-SY5Y cells after treatment with 5,10 a 20 µM CisPt for 24 h57 and in HepG2 cells treated with 25 µM CisPt for 12, 24 and 48 h58.
Capillary Western immunoassay was performed for detection of protein expression changes of PARP-1, JNK1 and JNK2. PARP-1 reflects the extent of DNA damage59 because it is cleaved by caspases 3/7 to two specific fragments of 24 and 89 kDa60. Thus, cleavage of PARP-1 protein by caspases is usually mentioned to be a hallmark of apoptosis61. Our results on detecting PARP-1 cleavage after treatment with 50 µM CisPt for 24 h are similar to the study of Ju et al.62 who detected the PARP-1 cleavage in HK-2 cells treated with 40 µM CisPt for 24 h. JNKs were reported to be strongly activated via phosphorylation in cells incubated with cisplatin63. In our study, expression of phosphorylated JNK isoforms (JNK1 and JNK2) was elevated after cisplatin treatment in all time intervals in dose-dependent manner compared to control cells which is similar to reported finding on increased pJNK expression in HeLa cells after incubation with 50 and 100 µM CisPt for 6 h64, and in pig kidney epithelial cells after 25 µM CisPt treatment for 24 h65.
Finally, TUNEL and DNA ladder assays were performed in CisPt treated cells. Both assays belong among frequently used methods for detection of DNA strand breaks and fragmentation14,45.
We found that TUNEL assay detected DNA fragmentation after 24 h of CisPt treatment. Thus, we obtained similar results to finding on DNA fragmentation detected using TUNEL assay in 50 µM CisPt treated HK-2 cells45 and LLC-PK1 cells55 after 24 h. The detection of internucleosomal DNA fragments using DNA ladder assay is in accordance with outcomes of studies of Qin et al. (2002) and Lau66 reporting occurrence of DNA ladder in CisPt treated HepG2 and LLC-PK1 cells after 24 and 48 h. We summarize that the findings of DNA fragmentation and nuclear condensation detected using all the biochemical assays in HepG2 and HK-2 cells after cisplatin treatment presented in this study are in accordance to published reports of other authors testing cisplatin as well.
Conclusion
We developed a spectrofluorometric method using Hoechst 33258 staining which detects nuclear condensation and fragmentation in intact cells. Our results showed that the spectrofluorometric method was capable to detect the nuclear changes in three typical pro-apoptotic agents, i.e. cisplatin, camptothecin, staurosporine, and in two cell lines of different origin. In addition, we compared the results obtained using the spectrofluorometric assay with outcomes of other methods characterizing apoptotic processes. Based on this comparison, we conclude that here developed spectrofluorometric method is capable to detect nuclear pro-apoptotic changes of similar sensitivity and specificity to that of caspase 3/7 activity measurement and TUNEL assay. Therefore, we suppose that the spectrofluorimetric H33258 method could join other routinely used methods characterizing apoptosis in cells. In comparison to TUNEL assay, the developed spectrofluorometric assay possesses several advantages (e.g. rapid processing, quantitative evaluation, low-cost) implying its potential use in assessing nuclear condensation and fragmentation in routine laboratory practice and in high-throughput screening studies.
Materials and methods
Chemicals
Hoechst 33258 solution (H33258, 1 mg/mL) and all other chemicals (formaldehyde, Triton X-100, BSA, NaCl, NaOH, Tris base, Na2EDTA·2H2O, PBS 10 ×, cisplatin, staurosporine, camptothecin, TiO2 P25) were purchased from Sigma Aldrich, USA.
Cell culture and treatment
All materials for cell culture were purchased from Sigma-Aldrich (USA) if not otherwise specified. HepG2 cells (ATCC HB-8065), a human hepatocellular carcinoma cell line, were purchased from ATCC (Manassas, VA, USA). HepG2 cells were cultured in Dulbecco's Modified Eagle's Medium with high glucose content (4500 g/L, w/wo phenol red) supplemented with 10% (v/v) fetal bovine serum, 50 μg/mL penicillin, 50 μg/mL streptomycin, 10 mM HEPES and 2 mM glutamine, maintained at 37 °C in a sterile, humidified atmosphere of 5% CO2. All the experiments were conducted using HepG2 cells in passages 4–15.
Human kidney, HK-2 cells (ATCC CRL-2190), a proximal tubular epithelial cell line derived from normal adult human kidney cells immortalized by transduction with human papillomavirus (HPV 16) DNA fragment67, were purchased from the ATCC (Manassas, VA, USA). The cells were cultured in supplemented Dulbecco’s modified Eagle’s medium (DMEM/F12 = 1:1) with 5% (v/v) fetal bovine serum, 1 mM pyruvate, 10 µg/mL insulin, 5.5 µg/mL transferrin, 5 ng/mL sodium selenite, 50 µg/mL penicillin, 50 µg/mL streptomycin, and 5 ng/mL epidermal growth factor according to a published protocol68,69. All the experiments were conducted using the HK-2 cells in passages 5–15.
HepG2 and HK-2 cells were tested for mycoplasma contamination using the MycoAlert Mycoplasma Detection Kit (Lonza). All cells used in the experiments were mycoplasma free. Short tandem repeat (STR) analysis (i.e., DNA fingerprinting) was used for HepG2 and HK-2 cell line authentication using a commercial kit in Generi Biotech (Czech Republic). The STR analysis proved 100% conformity of both HepG2 and HK-2 cells with the reference standards.
The HepG2 and HK-2 cells were seeded in 100 µL of appropriate cell culture medium in 96-well plates at density of 1.5 × 104 and 2 × 104 cells per well for 24 h (if not stated otherwise). To induce cell impairment, we used: cisplatin (CisPt, 0–100 µM), camptothecin (CAM; 0–5 µM), staurosporine (STA; 0–100 nM) and TiO2 P25 nanoparticles (NPs; 0–10 µg/mL). All compounds were diluted in appropriate cell culture mediums to obtain final concentrations. After seeding, the culture medium was replaced by 100 µL of medium containing a tested compound and the cells were treated for 6, 24 and 48 h. To characterize the extent of cell impairment, we used the newly developed spectrofluorometric assay using H33258 together with caspase activity and proteins expression measurements, TUNEL and DNA ladder assays.
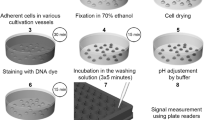
Hoechst 33258 spectrofluorometric assay
To develop a spectrofluorometric method for detection of changes in nuclear condensation and fragmentation in intact cells, we used a fluorescence dye H33258. To optimize the assay, we used HepG2 and HK-2 cells at confluency 50–70%. After treatment with tested compounds (CisPt, CAM, STA, NPs) for 6, 24 and 48 h, the cells grown in a 96-well plate were centrifuged (5 min, 8000g) at RT. Then, 70 µL of a supernatant was replaced with 70 µL of warmed phosphate-buffered saline (PBS 1 ×, 37 °C) and 10 µL of H33258 solution (in PBS 1 ×) was added to a well. The final concentrations of H33258 in a well were 0.1–5 µg/mL. Then, the cells were incubated with H33258 for 60 min during optimization of the assay, or for 5 min at optimal conditions and the spectrofluorometric measurement was performed at EX/EM = 352/461 nm (EX/EM slit widths 25/25 nm) using a Tecan Spark fluorescence microplate reader (Tecan, Switzerland) while incubated at 37 °C. The samples were measured at least in triplicates. After background subtraction, the fluorescence signal was presented in Relative Fluorescence Units (RFU) as mean ± SEM. All spectrofluorometric measurements presented here were repeated at least in three independent experiments.
Dehydrogenase activity measurement
Dehydrogenase activity was evaluated by WST-1 test (Roche, Germany). The WST-1 test measures the activity of intra- and extramitochondrial dehydrogenases. After cell treatment, 10 µL of WST-1 reagent was added to the treated cells. The absorbance change (0–1 h) was measured spectrophotometrically at wavelength of 440 nm using a Tecan Spark fluorescence microplate reader (Tecan, Switzerland). The cell viability was expressed as the percentage of intra- and extramitochondrial dehydrogenases activity relative to that in control cells (= 100%). The results were expressed as mean ± SD.
Glutathione assay
Glutathione levels were measured using the monochlorobimane spectrofluorometric assay70. After cell treatment, 20 µL of the bimane solution was added to cells to obtain the final concentration 40 µM and spectrofluorometric measurement was started immediately. The fluorescence increase (EX/EM = 394/490 nm) was measured for 10 min using a Tecan Spark fluorescence microplate reader (Tecan, Switzerland). The fluorescence was expressed as the slope of change in fluorescence over time. The glutathione levels were expressed as the percentage relative to the glutathione levels in control cells (= 100%). The results were expressed as mean ± SD.
Caspase 3/7 activity measurement
Caspase 3/7 activity in HepG2 and HK-2 cells was detected using ApoONE Homogeneous Caspase-3/7 Assay (Promega, USA) according to the manufacturer´s instructions. Briefly, 100 µL of the caspase 3/7 working solution was added to treated cells. After mixing, the cells were incubated for 30 min. Then, the fluorescence (EX/EM = 485/535 nm) was measured in duplicates using a Tecan Spark fluorescence microplate reader (Tecan, Switzerland) while incubated at 37 °C. The caspase 3/7 activity levels were expressed as the percentage relative to the caspase 3/7 activity levels in control cells (= 100%). The results were expressed as mean ± SEM.
Capillary Western Immunoassay
Capillary Western Immunoassay was performed in protein lysates from HepG2 and HK-2 cells cultured in 6-well plates at density of 5 × 105 and 1.3 × 106 cells per well, respectively. After seeding, the culture medium was replaced by 2 mL of CisPt solution and the cells were treated for 6, 24 and 48 h. After treatment, the cells were washed twice with PBS 1 × and protein lysates were prepared by lysing cells with 400 µL of RIPA buffer (Sigma-Aldrich, USA) with MS-safe protease and phosphatase inhibitor (Sigma-Aldrich, USA) on ice. Capillary Western Immunoassay was performed according to manufacturer´s instructions (Protein Simple, USA). Briefly, protein lysates were analyzed on a Wes system (ProteinSimple, USA) using a 12–230 kDa Separation Module (Biotechne, UK). Levels of phosphorylated JNK (pJNK, primary antibody 1:50, Promega, USA), poly-(ADP-ribose) polymerase-1 (PARP-1; primary antibody 1:100; Cell Signalling, USA) were normalized using the reference protein β-actin (primary antibody 1:500, Sigma-Aldrich, USA). The peaks were analyzed using Compass software (Protein Simple, USA). Two criteria were used for the discrimination of signals from the background: (1) the peak high must be higher or equal to 1000 and (2) the peak’s signal-to-noise ratio given by the software must be higher or equal to 10. The results were counted as:\(\frac{{{\text{area}}\,{\text{of}}\,{\text{the}}\,{\text{peak}}\,{\text{of}}\,{\text{interest}}}}{{{\text{area}}\,{\text{of}}\,{\text{the}}\,{\text{peak}}\,{\text{of}}\,\upbeta {\text{ - actin}}}}\).
TUNEL assay
TUNEL assay followed by H33258 staining of nuclei was performed in HepG2 and HK-2 cells cultured in 200 µL of appropriate cell culture medium on cell culture chamber slides at density of 1.5 × 105 and 2 × 105 cells per well, respectively. After seeding, the culture medium was replaced by 200 µL of CisPt solutions and the cells were treated for 6, 24 and 48 h. TUNEL assay was performed using Click-iT TUNEL Alexa Fluor 488 Imaging Assay kit (ThermoFisher Scientific, USA) according to manufacturer´s instructions. The cells were fixed with 12% formaldehyde for 15 min at 37 °C. Then, the cells were permeabilized with 0.2% Triton X-100 for 15 min at 37 °C, washed with PBS 1 × and incubated with terminal deoxynucleotidyl transferase (TdT) buffer for 10 min at 37 °C. After incubation, cells were mixed with a TdT reaction mixture (TdT buffer, 5-Ethynyl-2′-deoxyuridine 5′-triphosphate, TdT) and incubated for 1 h at 37 °C. Then, the cells were washed with 3% bovine serum albumin (BSA) and Click IT reagent for fluorescent staining was added for 30 min at 37 °C. After PBS 1 × washing, H33258 at a final concentration of 2 µg/mL was used to visualize the cell nuclei. DNA strand breaks (FITC filter, 480/30 nm) and cell nuclei (DAPI filter, 375/28 nm) were visualized with an Eclipse 80i fluorescence microscope (Nikon, Japan).
DNA ladder
DNA ladder was performed in HepG2 and HK-2 cells cultured in 6-well plates at density of 5 × 105 and 1 × 106 cells per well, respectively. After seeding, the culture medium was replaced by 2 mL of CisPt and the cells were treated for 6, 24 and 48 h. DNA was isolated from treated cells using The ApoTarget Quick Apoptotic DNA Ladder Detection Kit (Invitrogen, USA). Isolated DNA samples were loaded onto a 1.5% agarose gel with 0.5 mg/mL ethidium bromide (Top-Bio, Czech Republic) followed by electrophoresis (5 V/cm). Finally, DNA was visualized by an ultraviolet gel documentation system (Vilber Lourmat, Germany) at wavelength 254 nm. GeneRuler 100 bp DNA ladder (ThermoFisher Scientific, USA) was used as a DNA size standard.
Statistical analysis
Statistical analysis was performed using OriginPro 9.0.0 (OriginLab, USA). Statistical significance was analyzed after normality testing using one-way analysis of variance (ANOVA) followed by Tukey’s test at significance level p = 0.05 (*p < 0.05; **p < 0.01; ***p < 0.001).
Change history
03 September 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41598-021-97792-5
References
Wyllie, A. H. Glucocorticoid-induced thymocyte apoptosis is associated with endogenous endonuclease activation. Nature 284, 555–556. https://doi.org/10.1038/284555a0 (1980).
Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 35, 495–516. https://doi.org/10.1080/01926230701320337 (2007).
Caruso, S. et al. Defining the role of cytoskeletal components in the formation of apoptopodia and apoptotic bodies during apoptosis. Apoptosis 24, 862–877. https://doi.org/10.1007/s10495-019-01565-5 (2019).
Hou, L. et al. Necrotic pyknosis is a morphologically and biochemically distinct event from apoptotic pyknosis. J. Cell Sci. 129, 3084–3090. https://doi.org/10.1242/jcs.184374 (2016).
Burgoyne, L. A. The mechanisms of pyknosis: Hypercondensation and death. Exp. Cell Res. 248, 214–222. https://doi.org/10.1006/excr.1999.4406 (1999).
Gotzmann, J., Meissner, M. & Gerner, C. The fate of the nuclear matrix-associated-region-binding protein SATB1 during apoptosis. Cell Death Differ. 7, 425–438. https://doi.org/10.1038/sj.cdd.4400668 (2000).
Takada, S., Watanabe, T. & Mizuta, R. DNase gamma-dependent DNA fragmentation causes karyolysis in necrotic hepatocyte. J. Vet. Med. Sci. 82, 23–26. https://doi.org/10.1292/jvms.19-0499 (2020).
Ferri, K. F. & Kroemer, G. Control of apoptotic DNA degradation. Nat. Cell Biol. 2, E63-64. https://doi.org/10.1038/35008692 (2000).
Walsh, J. G. et al. Executioner caspase-3 and caspase-7 are functionally distinct proteases. Proc. Natl. Acad. Sci. USA 105, 12815–12819. https://doi.org/10.1073/pnas.0707715105 (2008).
Zhou, P., Lugovskoy, A. A., McCarty, J. S., Li, P. & Wagner, G. Solution structure of DFF40 and DFF45 N-terminal domain complex and mutual chaperone activity of DFF40 and DFF45. Proc. Natl. Acad. Sci. USA 98, 6051–6055. https://doi.org/10.1073/pnas.111145098 (2001).
Nagata, S., Nagase, H., Kawane, K., Mukae, N. & Fukuyama, H. Degradation of chromosomal DNA during apoptosis. Cell Death Differ. 10, 108–116. https://doi.org/10.1038/sj.cdd.4401161 (2003).
Skalka, M., Matyasova, J. & Cejkova, M. Dna in chromatin of irradiated lymphoid tissues degrades in vivo into regular fragments. FEBS Lett. 72, 271–274. https://doi.org/10.1016/0014-5793(76)80984-2 (1976).
Pariente, R., Pariente, J. A., Rodríguez, A. B. & Espino, J. Melatonin sensitizes human cervical cancer HeLa cells to cisplatin-induced cytotoxicity and apoptosis: Effects on oxidative stress and DNA fragmentation. J. Pineal Res. 60, 55–64. https://doi.org/10.1111/jpi.12288 (2016).
Majtnerová, P. & Roušar, T. An overview of apoptosis assays detecting DNA fragmentation. Mol. Biol. Rep. 45, 1469–1478. https://doi.org/10.1007/s11033-018-4258-9 (2018).
Martin, R. M., Leonhardt, H. & Cardoso, M. C. DNA labeling in living cells. Cytometry A 67, 45–52. https://doi.org/10.1002/cyto.a.20172 (2005).
Zhang, X. T., Song, T. B., Du, B. L., Li, D. M. & Li, X. M. Caspase-3 antisense oligodeoxynucleotides inhibit apoptosis in gamma-irradiated human leukemia HL-60 cells. Apoptosis 12, 743–751. https://doi.org/10.1007/s10495-006-0018-8 (2007).
Miao, Z. H. et al. 4-nitroquinoline-1-oxide induces the formation of cellular topoisomerase I-DNA cleavage complexes. Cancer Res. 66, 6540–6545. https://doi.org/10.1158/0008-5472.CAN-05-4471 (2006).
Seiler, J. A., Syed, A., Aladjem, M. I. & Pommier, Y. Replication checkpoint selectivity for late S phase cells induced by topoisomerase I cleavage complexes. Cancer Res. 66, 1153 (2006).
Gomes, C. J., Harman, M. W., Centuori, S. M., Wolgemuth, C. W. & Martinez, J. D. Measuring DNA content in live cells by fluorescence microscopy. Cell Div. 13, 6. https://doi.org/10.1186/s13008-018-0039-z (2018).
Liu, C.-H., Tsao, M.-H., Sahoo, S. L. & Wu, W.-C. Magnetic nanoparticles with fluorescence and affinity for DNA sensing and nucleus staining. RSC Adv. 7, 5937–5947. https://doi.org/10.1039/c6ra25610d (2017).
Carlson, C. R., Grallert, B., Bernander, R., Stokke, T. & Boye, E. Measurement of nuclear DNA content in fission yeast by flow cytometry. Yeast 13, 1329–1335. https://doi.org/10.1002/(SICI)1097-0061(199711)13:14%3c1329::AID-YEA185%3e3.0.CO;2-M (1997).
Vardevanyan, P. O., Parsadanyan, M. A., Antonyan, A. P., Shahinyan, M. A. & Karapetyan, A. T. Spectroscopic study of interaction of various GC-content DNA with Hoechst 33258 depending on Na(+) concentration. J. Biomol. Struct. Dyn. https://doi.org/10.1080/07391102.2020.1730244 (2020).
Campos, V., Rappaz, B., Kuttler, F., Turcatti, G. & Naveiras, O. High-throughput, nonperturbing quantification of lipid droplets with digital holographic microscopy. J. Lipid Res. 59, 1301–1310. https://doi.org/10.1194/jlr.D085217 (2018).
Nogueira, E. et al. Assessment of liposome disruption to quantify drug delivery in vitro. Biochim. Biophys. Acta 1858, 163–167. https://doi.org/10.1016/j.bbamem.2015.11.008 (2016).
Zhou, Y. B. et al. Multiplexed imaging detection of live cell intracellular changes in early apoptosis with aggregation-induced emission fluorogens. Sci. China Chem. 61, 892–897. https://doi.org/10.1007/s11426-018-9287-x (2018).
Sakamoto, T., Hasegawa, D. & Fujimoto, K. Fluorine-modified bisbenzimide derivative as a molecular probe for bimodal and simultaneous detection of DNAs by F-19 NMR and fluorescence. Chem. Commun. 51, 8749–8752. https://doi.org/10.1039/c5cc01995h (2015).
Martin, R. M., Leonhardt, H. & Cardoso, M. C. DNA labeling in living cells. Cytom. Part A 67A, 45–52. https://doi.org/10.1002/cyto.a.20172 (2005).
Qin, G. Q. et al. Dihydroartemisinin induces apoptosis preferentially via a Bim-mediated intrinsic pathway in hepatocarcinoma cells. Apoptosis 20, 1072–1086. https://doi.org/10.1007/s10495-015-1132-2 (2015).
Kapoor, R., Rizvi, F. & Kakkar, P. Naringenin prevents high glucose-induced mitochondria-mediated apoptosis involving AIF, Endo-G and caspases. Apoptosis 18, 9–27. https://doi.org/10.1007/s10495-012-0781-7 (2013).
Chen, G. et al. Mammalian target of rapamycin regulates isoliquiritigenin-induced autophagic and apoptotic cell death in adenoid cystic carcinoma cells. Apoptosis 17, 90–101. https://doi.org/10.1007/s10495-011-0658-1 (2012).
Soares, J. et al. Structure-cytotoxicity relationship profile of 13 synthetic cathinones in differentiated human SH-SY5Y neuronal cells. Neurotoxicology 75, 158–173. https://doi.org/10.1016/j.neuro.2019.08.009 (2019).
Zhang, Z. et al. Cytochrome P450 2A13 is an efficient enzyme in metabolic activation of aflatoxin G1 in human bronchial epithelial cells. Arch. Toxicol. 87, 1697–1707. https://doi.org/10.1007/s00204-013-1108-3 (2013).
Wang, J. et al. Biochanin A protects against lipopolysaccharide-induced damage of dopaminergic neurons both in vivo and in vitro via inhibition of microglial activation. Neurotox. Res. 30, 486–498. https://doi.org/10.1007/s12640-016-9648-y (2016).
Ki, S. H. et al. Artificial vesicles as an animal cell model for the study of biological application of non-thermal plasma. J. Phys. D Appl. Phys. 49, Artn 085401 https://doi.org/10.1088/0022-3727/49/8/085401 (2016).
Hadi, L. M., Yaghini, E., Stamati, K., Loizidou, M. & MacRobert, A. J. Therapeutic enhancement of a cytotoxic agent using photochemical internalisation in 3D compressed collagen constructs of ovarian cancer. Acta Biomater. 81, 80–92. https://doi.org/10.1016/j.actbio.2018.09.041 (2018).
Aston, N. S., Watt, N., Morton, I. E., Tanner, M. S. & Evans, G. S. Copper toxicity affects proliferation and viability of human hepatoma cells (HepG2 line). Hum. Exp. Toxicol. 19, 367–376. https://doi.org/10.1191/096032700678815963 (2000).
Handl, J., Capek, J., Majtnerova, P., Bacova, J. & Rousar, T. The effect of repeated passaging on the susceptibility of human proximal tubular HK-2 cells to toxic compounds. Physiol. Res. 69, 731–738. https://doi.org/10.33549/physiolres.934491 (2020).
Siemann, D. W. & Keng, P. C. Cell cycle specific toxicity of the Hoechst 33342 stain in untreated or irradiated murine tumor cells. Cancer Res. 46, 3556–3559 (1986).
Purschke, M., Rubio, N., Held, K. D. & Redmond, R. W. Phototoxicity of Hoechst 33342 in time-lapse fluorescence microscopy. Photochem. Photobiol. Sci. 9, 1634–1639. https://doi.org/10.1039/c0pp00234h (2010).
Hammond, C. L., Madejczyk, M. S. & Ballatori, N. Activation of plasma membrane reduced glutathione transport in death receptor apoptosis of HepG2 cells. Toxicol. Appl. Pharmacol. 195, 12–22. https://doi.org/10.1016/j.taap.2003.10.008 (2004).
Cao, M. R. et al. Harmine induces apoptosis in HepG2 cells via mitochondrial signaling pathway. Hepatobiliary Pancreat. Dis. Int. 10, 599–604. https://doi.org/10.1016/s1499-3872(11)60102-1 (2011).
Beaton-Green, L. A. & Wilkins, R. C. Quantitation of chromosome damage by imaging flow cytometry. Methods Mol. Biol. 1389, 97–110. https://doi.org/10.1007/978-1-4939-3302-0_6 (2016).
Bogush, T. A. et al. A new approach to epithelial-mesenchymal transition diagnostics in epithelial tumors: Double immunofluorescent staining and flow cytometry. Biotechniques https://doi.org/10.2144/btn-2020-0024 (2020).
Zhang, L. J., Li, Z. Q., Yang, Y. P., Li, X. W. & Ji, J. F. Tunicamycin suppresses cisplatin-induced HepG2 cell apoptosis via enhancing p53 protein nuclear export. Mol. Cell. Biochem. 327, 171–182. https://doi.org/10.1007/s11010-009-0055-z (2009).
Yang, S. K. et al. Mitochondria targeted peptide SS-31 prevent on cisplatin-induced acute kidney injury via regulating mitochondrial ROS-NLRP3 pathway. Biomed. Pharmacother. 130, 110521. https://doi.org/10.1016/j.biopha.2020.110521 (2020).
Tanida, S. et al. Mechanisms of cisplatin-induced apoptosis and of cisplatin sensitivity: Potential of BIN1 to act as a potent predictor of cisplatin sensitivity in gastric cancer treatment. Int. J. Surg. Oncol. 2012, 862879. https://doi.org/10.1155/2012/862879 (2012).
Garcia, C. P. et al. Topoisomerase I inhibitor, camptothecin, induces apoptogenic signaling in human embryonic stem cells. Stem Cell Res. 12, 400–414. https://doi.org/10.1016/j.scr.2013.12.002 (2014).
Rath, G. et al. De novo ceramide synthesis is responsible for the anti-tumor properties of camptothecin and doxorubicin in follicular thyroid carcinoma. Int. J. Biochem. Cell B 41, 1165–1172. https://doi.org/10.1016/j.biocel.2008.10.021 (2009).
Pommier, Y. Topoisomerase I inhibitors: Camptothecins and beyond. Nat. Rev. Cancer 6, 789–802. https://doi.org/10.1038/Nrc1977 (2006).
Ding, Y. M., Wang, B., Chen, X. Y., Zhou, Y. & Ge, J. H. Staurosporine suppresses survival of HepG2 cancer cells through Omi/HtrA2-mediated inhibition of PI3K/Akt signaling pathway. Tumor Biol. 39, Artn 694317 https://doi.org/10.1177/1010428317694317 (2017).
Deshmukh, M. & Johnson, E. M. Staurosporine-induced neuronal death: Multiple mechanisms and methodological implications. Cell Death Differ. 7, 250–261. https://doi.org/10.1038/sj.cdd.4400641 (2000).
Petkovic, J. et al. DNA damage and alterations in expression of DNA damage responsive genes induced by TiO2 nanoparticles in human hepatoma HepG2 cells. Nanotoxicology 5, 341–353. https://doi.org/10.3109/17435390.2010.507316 (2011).
Ji, Y. B. & Gao, S. Y. Arylamine N-acetyltransferases: a new inhibitor of apoptosis in HepG2 cells. J. Zhejiang Univ.-Sc B 9, 701–706. https://doi.org/10.1631/jzus.B0820090 (2008).
Gorman, A. M., Hirt, U. A., Orrenius, S. & Ceccatelli, S. Dexamethasone pre-treatment interferes with apoptotic death in glioma cells. Neuroscience 96, 417–425. https://doi.org/10.1016/S0306-4522(99)00565-5 (2000).
Park, M. S., De Leon, M. & Devarajan, P. Cisplatin induces apoptosis in LLC-PK1 cells via activation of mitochondrial pathways. J. Am. Soc. Nephrol. 13, 858–865 (2002).
Schweyer, S. et al. Cisplatin-induced apoptosis in human malignant testicular germ cell lines depends on MEK/ERK activation. Br. J. Cancer 91, 589–598. https://doi.org/10.1038/sj.bjc.6601919 (2004).
Rathinam, R., Ghosh, S., Neumann, W. L. & Jamesdaniel, S. Cisplatin-induced apoptosis in auditory, renal, and neuronal cells is associated with nitration and downregulation of LMO4. Cell Death Discov. 1, Artn 15052 https://doi.org/10.1038/Cddiscovery.2015.52 (2015).
Yasuda, M., Kishimoto, S., Amano, M. & Fukushima, S. The involvement of pregnane X receptor-regulated pathways in the antitumor activity of cisplatin. Anticancer Res. 39, 3601–3608. https://doi.org/10.21873/anticanres.13507 (2019).
Chaudhuri, A. R. & Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 18, 610–621. https://doi.org/10.1038/nrm.2017.53 (2017).
Chaitanya, G. V., Steven, A. J. & Babu, P. P. PARP-1 cleavage fragments: Signatures of cell-death proteases in neurodegeneration. Cell Commun. Signal. 8, Artn 31 https://doi.org/10.1186/1478-811x-8-31 (2010).
Kaufmann, S. H., Desnoyers, S., Ottaviano, Y., Davidson, N. E. & Poirier, G. G. Specific proteolytic cleavage of poly(adp-ribose) polymerase—An early marker of chemotherapy-induced apoptosis. Cancer Res. 53, 3976–3985 (1993).
Ju, S. M. et al. Phosphorylation of eIF2 alpha suppresses cisplatin-induced p53 activation and apoptosis by attenuating oxidative stress via ATF4-mediated HO-1 expression in human renal proximal tubular cells. Int. J. Mol. Med. 40, 1957–1964. https://doi.org/10.3892/ijmm.2017.3181 (2017).
Hayakawa, J., Depatie, C., Ohmichi, M. & Mercola, D. The activation of c-Jun NH2-terminal kinase (JNK) by DNA-damaging agents serves to promote drug resistance via activating transcription factor 2 (ATF2)-dependent enhanced DNA repair. J. Biol. Chem. 278, 20582–20592. https://doi.org/10.1074/jbc.M210992200 (2003).
Brozovic, A. et al. Long-term activation of SAPK/JNK, p38 kinase and Fas-L expression by cisplatin is attenuated in human carcinoma cells that acquired drug resistance. Int. J. Cancer 112, 974–985. https://doi.org/10.1002/ijc.20522 (2004).
Lee, D., Lee, S. R., Kang, K. S. & Kim, K. H. Benzyl salicylate from the stems and stem barks of Cornus walteri as a nephroprotective agent against cisplatin-induced apoptotic cell death in LLC-PK1 cells. RSC Adv. 10, 5777–5784. https://doi.org/10.1039/c9ra07009e (2020).
Lau, A. H. Apoptosis induced by cisplatin nephrotoxic injury. Kidney Int. 56, 1295–1298. https://doi.org/10.1046/j.1523-1755.1999.00687.x (1999).
Ryan, M. J. et al. HK-2: An immortalized proximal tubule epithelial cell line from normal adult human kidney. Kidney Int. 45, 48–57. https://doi.org/10.1038/ki.1994.6 (1994).
Hauschke, M. et al. Neutrophil gelatinase-associated lipocalin production negatively correlates with HK-2 cell impairment: Evaluation of NGAL as a marker of toxicity in HK-2 cells. Toxicol. In Vitro 39, 52–57. https://doi.org/10.1016/j.tiv.2016.11.012 (2017).
Handl, J. et al. Transient increase in cellular dehydrogenase activity after cadmium treatment precedes enhanced production of reactive oxygen species in human proximal tubular kidney cells. Physiol. Res. 68, 481–490. https://doi.org/10.33549/physiolres.934121 (2019).
Capek, J., Hauschke, M., Bruckova, L. & Rousar, T. Comparison of glutathione levels measured using optimized monochlorobimane assay with those from ortho-phthalaldehyde assay in intact cells. J. Pharmacol. Tox. Met. 88, 40–45. https://doi.org/10.1016/j.vascn.2017.06.001 (2017).
Funding
The financial support was received from the Ministry of Education, Youth and Sports of the Czech Republic via projects NANOBIO (Reg. No. CZ.02.1.01/0.0/0.0/17_048/0007421).
Author information
Authors and Affiliations
Contributions
Cell culture and treatment, J.H. and P.M., spectrofluorometric measurements, P.M. and J.C.; TUNEL, J.C.; DNA ladder, P.M.; protein analysis, P.M.; and F.P.; manuscript preparation, P.M. and T.R.; editing; J.C., J.H.; supervision; T.R. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this Article was revised: The original version of this Article contained an error in the spelling of the authors Pavlina Majtnerova, Jan Capek, Filip Petira, Jiri Handl and Tomas Rousar which were incorrectly given as Majtnerova Pavlina, Capek Jan, Petira Filip, Handl Jiri and Rousar Tomas respectively.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Majtnerova, P., Capek, J., Petira, F. et al. Quantitative spectrofluorometric assay detecting nuclear condensation and fragmentation in intact cells. Sci Rep 11, 11921 (2021). https://doi.org/10.1038/s41598-021-91380-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-91380-3
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
