
Abstract
Postaxial polydactyly is characterised by fifth digit duplications in hands and/or feet. Two phenotypic varieties have been described. In type A, the extra digit is well formed and articulates with the fifth or an extra metacarpal. A rudimentary extra fifth digit characterises type B. Mutations in the GLI3 gene are associated with postaxial polydactyly in some families and a second locus has been identified on chromosome 13 but the majority of cases remain unexplained. We report here a third locus for postaxial polydactyly on chromosome 19p13.1-13.2 in a large Chinese kindred with a combination of type A and B. Two-point linkage analysis showed the highest LOD score (Zmax = 5.85 at θ = 0 cM) at marker D19S221. Recombination events with markers D19S1165 and D19S929 define the critical region for this postaxial polydactyly locus to a region of 4.3 cM on the genetic map.
Similar content being viewed by others

Introduction
Postaxial polydactyly (PAP) is a frequent congenital hand malformation characterized by fifth digit duplications in hands and/or feet. The prevalence estimates range from 1/630 to 1/3300 in Caucasians and from 1/100 to 1/300 in Blacks.1
Two phenotypic varieties have been described, in type A (PAP-A); the extra digit is well formed and articulates with the fifth or an extra metacarpal. Type B (PAP-B) is characterised by a rudimentary extra fifth digit (pedunculated postminimus) and is usually represented by an extra skin tag. Estimates for the penetrance values for PAP-A vary between 63 and 100% and for PAP-B around 48%.2,3,4
PAP can segregate as an autosomal dominant or recessive trait, and is often observed in association with other syndromes.5,6,7,8,9,10 Partial cutaneous syndactyly of the 4th and 5th digit is often seen in combination with PAP. Genetic studies on families with isolated PAP have revealed at least two autosomal dominant loci responsible for the phenotype. In a large Indian kindred with PAP-A with complete penetrance, Radhakrishna et al.11 found a frameshift mutation in the Gli3 gene on chromosome 7 (PAPA1) [MIM 174200]. A second candidate region for PAP was indicated by the observation that PAP occurs in approximately 75% of trisomy 13 cases.12 Subsequently, a locus for an autosomal dominant PAP-A with reduced penetrance was mapped to chromosome 13q in a large Turkish kindred (PAPA2) [MIM 602085].10 PAP-A and B sometimes occur in the same family and it has been suggested that a single gene might be responsible for both types of polydactyly.13,14,15,16 Recently this suggestion was confirmed by the finding that mutations in GLI3 are associated with PAP-A and B within a single family.17
In this study we describe a large kindred from the northern part of China, with a combination of autosomal dominant PAP-A and B with incomplete penetrance. Linkage of the phenotype to chromosomes 7 and 13q could be excluded. Subsequently a genome wide search with highly informative polymorphic markers was performed and strong evidence for linkage was found on chromosome 19p. Our findings demonstrate further genetic heterogeneity for PAP and this third locus shows clinical variation in that both PAP-A and B are observed in members from a single family.
Material and methods
The study was approved by the authorities of Peking Union Medical College and the Medical Ethical Committee of the Erasmus University Rotterdam. All blood samples were collected with signed informed consent (written in Chinese) stating the subsequent use of their blood samples for scientific research use only.
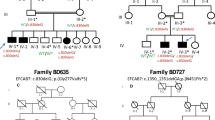
A large six generation kindred was ascertained from the province of Hebei in the northern part of China, with a combination of autosomal-dominant PAP-A and B with incomplete penetrance (Figure 1).18 Consanguinity was not observed within the pedigree. Blood samples were collected from 36 individuals, with seven affected males and 10 affected females with PAP-A and/or B. Clinical diagnosis was based on observation by one of us, or on family history, because surgical corrections were performed on most patients shortly after birth. Physical examinations of the patients included cardiac percussion and stethoscope auscultation. No additional symptoms were detected and none of the patients had a history of cardiac and/or renal disease.

Pedigree of the Chinese family with a combination of autosomal dominant PAP-A and -B with incomplete penetrance. Blackened symbols denote affected individuals, and symbols with slashes denote deceased individuals. A symbol with a black vertical bar indicates affected by family history. Question marks indicate failed genotypings.
Genomic DNA was isolated from peripheral blood by a simple salting out protocol.19 Microsatellite markers from the CHLC/Weber Human Screening Set V5 (n = 363) or obtained from the Genome database (n = 6) were tested essentially as described by Weber and May.20 The genome screen was performed on 28 DNA samples.
LOD scores were calculated using the MLINK and LINKMAP programs of the LINKAGE package (version 5.1).21 PAP was assumed to be an autosomal dominant disease with a gene frequency of 0.001 and a penetrance value of 95%. The phenocopy rate was set at zero. The mutation rate was set at zero and equal recombination rates between males and females were assumed. Because of the lack of data on marker allele frequencies from the Chinese population, marker allele frequencies were calculated from independent individuals from the PAP family. Calculation of pairwise LOD scores with equal allele frequencies did not substantially alter results (<10%).
URLs for data in this report are as follows: Online Mendelian Inheritance in Man (OMIM, http://www.ncbi.nlm.nih.gov/Omim/) for PAPA1 [MIM 174200] PAPA2 [MIM 602085]. Center for Medical Genetics, Marshfield Medical Research Foundation (http://research.marshfieldclinic.org/genetics/) for information on polymorphic markers and localization of polymorphic markers. The Genome Database is available through http://gdbwww.gdb.org/.Human Genome Center at Lawrence Livermore National Library (http://bbrp.llnl.gov/genome/html/chrom_map.html) for information on physical map of chromosome 19. Ensembl database (http://www.ensembl.org) and the Human Genome Browser (http://genome.cse.ucsc.edu/) for identification of candidate genes.
Results and discussion
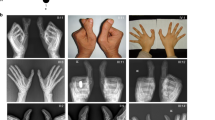
Clinical data were available from 16 affected individuals (Table 1). Most affected individuals showed well developed and functional extra postaxial digits on hands and/or feet, but the expression of the phenotype is variable (Figure 2); all affected individuals show PAP-A of the feet; fourteen individuals bilateral and two individuals unilateral. Foot X-rays were available for individual V-6 (Figure 2d).

Clinical phenotype of patients from the PAP-3 family. (a) Hands of III-1. (b) Hands of V-11. (c) Feet of V-6. (d) X-ray of feet V-6.
Seven affected individuals showed polydactyly on the hands as well. Four of them showed PAP-A on both hands. One individual had PAP-B on both hands and two individuals presented with PAP-A on one hand and PAP-B on the other hand (Table 1). No syndactyly of hand or feet and no other associated anomalies were observed in this family. One female individual has a normal phenotype but her two daughters are affected, demonstrating that she most likely is an obligate carrier. X rays for her were not available.
For our linkage studies we initially tested markers from chromosome 7p and 13q where two other PAP loci have been mapped but found significant negative LOD scores for all markers studied (data not shown). We then set out to perform a genome wide screen with highly polymorphic markers. We found significant positive LOD scores for markers on chromosome 19p (Table 2). The highest LOD score was obtained with D19S221 (Zmax = 5.85 at θ = 0 cM). Significant positive LOD scores were obtained for no other marker . This is the third PAP locus and we propose to call it PAP3.
Haplotype analysis of chromosome 19 markers using the marker order from the CHLC marker map or the Marshmed marker map caused several double recombination events in individuals from the pedigree indicating that the marker order might be incorrect. These double recombination events were not observed if markers were ordered according to the data from the Human Genome draft sequence. Therefore the marker order from the Human Genome draft sequence was used for haplotype analysis: D19S583–D19S394–D19S584–D19S1165–D19S221–D19S558–D19S840–D19S226–D19S929–D19S252.
We found a common haplotype for all patients for markers D19S221, D19S558, D19S840 and D19S226, listed from the telomeric to the centromic side (Figure 1). The first recombination event on the telomeric side is found between marker D19S221 and D19S1165 (individual IV-15). On the centromeric side the first recombination event is observed between marker D19S226 and D19S929 (individual V-10). Therefore the critical region for PAP3 is located between markers D19S1165 and D19S929, a region of 4.3 cM on the genetic map or about 2.5 Mb on the physical map. On the cytogenetic map this region is located at chromosome bands 19p13.2–13.1. Unaffected individual V-1 could reduce this region due to a recombination event between marker D19S840 and D19S226 however because the uncertainty of the penetrance for PAP it is not clear if this person is a non-carrier or a case of non-penetrance.
In our family only a single individual out of nine transmitting individuals (89%) showed no clinical phenotype. Compared to other studies were penetrance values for PAP-A varies between 63 and 100% and for PAP-B around 48%2,3,4 the penetrance was high in this family. However, there is a large variation in the expression of the phenotype. Whether additional genetic factors or environmental factors cause this variation is currently not known.
The critical region for PAP3 harbours around 60 known genes. Most of the genes are housekeeping genes that have a variety of functions in cells. It also contains a number of interesting candidate genes. A cluster of zinc fingers containing genes are present in the region which can be considered candidate genes for PAP because zinc finger containing genes often act as transcription factors and several developmental disorders are associated with mutations in transcription factors. For example, GLI3, the gene associated with Greig syndrome, preaxial polydactyly type IV and PAP-A/B is a zinc finger containing gene of the Kruppel family. Therefore the Kruppel like factor 1 (KLF1) and the gonadotropin inducible transcription repressor-2 (GIOT-2) were considered strong candidate genes. The epithelial growth factor domains containing EMR2 protein has structural features characteristic of extracellular matrix proteins and transmembrane proteins suggesting that it is a receptor involved in both cell adhesion and signaling and was therefore also considered a candidate gene.22 However, no mutations were identified in patients in the coding sequences plus intron-exon boundaries of these genes. There are several other zinc finger genes such as KIAA1198 and KIAA0973 mapped in this area. Although their function and expression pattern is poorly characterized they might also be considered candidate genes.
The homologous region in mouse of this part of human chromosome 19 is located on chromosome 8. We have searched the Mouse Genome Database for mouse limb phenotypes that could serve as model systems. Interestingly, the mouse oligosyndactylism (Os) has been localized in this region. Although in our family no syndactyly was observed, cutaneous syndactyly is not uncommon in polydactyly families. The mouse mutation is X-ray induced, which often results in large genomic deletions. It is possible that much milder mutation would cause PAP. Positional cloning of either the gene responsible for human PAP3 or the mouse Os phenotype will clarify this.
References
Temtamy SA . Polydactyly, postaxial in Buyse ML ed Birth defects encyclopedia Cambridge, MA: Blackwell Scientific 1990 pp 1397–1398
Temtamy SA, McKusick VA . Synopsis of hand malformation with particular emphasis on genetic factors Birth Defects 1969 5: 125–184
Castilla E, Paz JE, Mutchinick O, Munoz E, Giorgiutti E, Gelman Z . Polydactyly: a genetic study in South America Am J Hum Genet 1973 25: 405–412
Scott-Emuakpor AB, Madueke EDN . The study of genetic variation in Nigeria. II. The genetics of polydactyly Hum Hered 1976 26: 198–202
Mohan J . Postaxial polydactyly in three Indian families J Med Genet 1969 6: 196–200
Cantu JM, del Castillo V, Cortes R, Urrusti J . Autosomal recessive postaxial polydactyly: report of a family Birth Defects Orig Artic Ser 1974 10: 19–22
Mollica F, Volti SL, Sorge G . Autosomal recessive postaxial polydactyly type A in a Sicilian family J Med Genet 1978 15: 212–216
Merlob P, Grunebaum M, Reisner SH . A newborn infant with craniofacial dysmorphism and polysyndactyly (Greig's syndrome) Acta Paediatr Scand 1981 70: 275–277
Radhakrishna U, Blouin J-L, Mehenni H et al. Mapping one form of autosomal dominant postaxial polydactyly type A to chromosome 7p15-q11.23 by linkage analysis Am J Hum Genet 1997 60: 597–604
Akarsu AN, Ozbas F, Kostakoglu N . Mapping of the second locus of postaxial polydactyly type A (PAP-A2) to chromosome 13q21-q32 Am J Hum Genet 1997 61 (suppl.): A265
Radhakrishna U, Wild A, Grzeschik KH, Antonarakis SE . Mutation in GLI3 in postaxial polydactyly type A Nat Genet 1997 17: 269–271
Lewandowski RC Jr, Yunis JJ . Phenotypic mapping in man in: Yunis JJ New Chromosomal Syndromes Academic Press: New York 1977 pp 369–394
Sverdrup A . Postaxial polydactylism in six generations of a Norwegian family J Genet 1922 12: 217–240
Odiorne JM . Polydactylism in related New England families J Hered 1943 34: 45–56
Ventruto V, Theo G, Celona A et al. A and B postaxial polydactyly in two members of the same family Clin Genet 1980 18: 342–347
Kucheria K, Kenue RK, Taneja N . An Indian family with postaxial polydactyly in four generations Clin Genet 1981 20: 36–39
Radhakrishna U, Bornholdt D, Scott HS et al. The phenotypic spectrum of GLI3 morphopathies includes autosomal dominant preaxial polydactyly type-IV and postaxial polydactyly type-A/B; No phenotype prediction from the position of GLI3 mutations Am J Hum Genet 1999 65: 645–655
Hongshan Zhao, Shnagzhi Huang, Ying Zou, Jinghua Chai, Hui Li, Mingyue Li . Exclusive mapping on polydactyly with markers on chromosomes 7 and 2 in a Chinese kindred Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2000 17: 6–9
Miller SA, Dykes DD, Polesky HF . A simple salting out procedure for extracting DNA from human nucleated cells Nucl Acids Res 1988 16: 1215
Weber JL, May PE . Abundant class of human DNA polymorphisms which can be typed using the polymerase chain reaction Am J Hum Genet 1989 44: 388–396
Lathrop GM, Lalouel JM . Easy calculations of lodscores and genetic risks on a small computer Am J Hum Genet 1984 36: 460–465
Hsi-Hsien Lin, Martin S, Jorg H, Siamon G, Andrew JM . Human EMR2, a novel EGF-TM7 molecule on chromosome 19p13.1, is closely related to CD97 Genomics 2000 67: 188–200
Acknowledgements
The authors are grateful for the dedicated cooperation of the family members participating in this study. The authors wish to thank Prof Hans Galjaard for support and the ‘Stichting Klinische Genetica’ for financial support. This work was also supported in part by the grant from National High Technology Research and Development Program (Z19-01-04-01), People's Republic of China.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zhao, H., Tian, Y., Breedveld, G. et al. Postaxial polydactyly type A/B (PAP-A/B) is linked to chromosome 19p13.1-13.2 in a chinese kindred. Eur J Hum Genet 10, 162–166 (2002). https://doi.org/10.1038/sj.ejhg.5200790
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5200790
Keywords
This article is cited by
-
The molecular genetics of human appendicular skeleton
Molecular Genetics and Genomics (2022)
-
Exome sequencing revealed a splice site variant in the IQCE gene underlying post-axial polydactyly type A restricted to lower limb
European Journal of Human Genetics (2017)
-
Genetic mapping of an autosomal recessive postaxial polydactyly type A to chromosome 13q13.3–q21.2 and screening of the candidate genes
Human Genetics (2012)
-
A new locus for postaxial polydactyly type A/B on chromosome 7q21–q34
European Journal of Human Genetics (2003)
