
Abstract
Identification and characterization of several important regulators of angiogenesis, and FDA approval of the first antiangiogenic drugs, has opened a new era in the therapy of cancer and neovascular age-related macular degeneration. This brief review focuses on the progress in targeting one of the major regulators of angiogenesis, VEGF-A, and also discusses potential cellular and molecular mechanisms underlying resistance to antiangiogenic treatments.
Similar content being viewed by others

Physiological and pathological angiogenesis
Angiogenesis is a fundamental process occurring during embryonic and adult life, resulting in the formation of new blood vessels. Physiological angiogenesis, which requires the coordinated action of a variety of ligands and receptors on endothelial and mural cells, is essential for tissue maintenance and homeostasis.1 There are several positive and negative regulators of angiogenesis. Vascular endothelial growth factor (VEGF), angiopoietins, members of the FGF family are among the positive regulators, whereas IL-12, thrombospondin, angiostatin and endostatin are inhibitors of angiogenesis.
Similar to normal tissues, solid tumors require new blood vessels for growth and survival. In addition, neovascularization is a prominent features of several intraocular neovascular syndromes, including the wet form of age-related macular degeneration (AMD), the leading cause of blindness in the elderly.2 Therefore, antiangiogenic agents are attractive candidates for the treatment of several disorders.
VEGF-A: a key regulator of angiogenesis
There is much evidence that VEGF is a key regulator of developmental angiogenesis as loss of a single VEGF allele results in embryonic lethality.3 The VEGF pathway also plays essential role in reproductive and bone angiogenesis. In mammals, the VEGF family comprises of five members including VEGF-A (thereafter called VEGF), VEGF-B, VEGF-C, VEGF-D and PlGF (placenta growth factor). Alternative exon splicing results in generation of several VEGF isoforms including VEGF121, VEGF165, VEGF189 and VEGF206. Furthermore, plasmin and various metalloproteinases can cleave VEGF165 at the COOH terminus, generating bioactive nonheparin-binding fragments.3
Three tyrosine kinase receptors bind members of the VEGF gene family: VEGFR-1 (Flt-1), VEGFR-2 (KDR) and VEGFR-3. Moreover, co-receptors, such as heparan sulfate proteoglycans and neuropilins, may facilitate activation of VEGFRs (reviewed by Ferrara et al3). Members of the VEGF gene family show different affinities for one of the three receptors. VEGF-B and PlGF bind selectively to VEGFR-1. VEGF is the main ligand for VEGFR-2 but proteolytically cleaved forms of VEGF-C and VEGF-D may also bind and activate this receptor. Finally, VEGFR-3 is activated only by VEGF-C and VEGF-D. VEGFR-1 and VEGFR-2 are expressed in vascular endothelial cells, monocytes, macrophages and hematopoietic stem cells (HSCs). VEGFR-1 is also expressed in certain nonendothelial cell types.3 Interestingly, subsets of liquid and solid tumor cells were found to express VEGFR-1 and VEGFR-2.4 In contrast to VEGFR-1 and -2, VEGFR-3 is involved in the regulation of lymphangiogenesis and its expression in the adult appears to be largely restricted to lymphatic endothelial cells.5
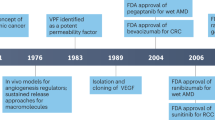
Clinical development of anti-VEGF agents
The existence of numerous angiogenic factors, including EGF, TGF-α TGF-β, acidic and basic FGF (reviewed by Ferrara and Kerbel6), suggested the contribution of multiple factors to tumor angiogenesis. Therefore, blocking a single angiogenic molecule was expected to have little or no impact on tumor growth. However, in apparent contrast with this view, experiments with neutralizing antibodies and other inhibitors demonstrated that blockade of VEGF alone can substantially suppress tumor growth and angiogenesis in several models.7 These encouraging findings prompted efforts for the development of therapies aimed at targeting VEGF and several pharmacologic approaches have been developed to inhibit the VEGF axis, based on targeting the ligands (mainly VEGF) or the receptors (VEGFR-1 and VEGFR-2).3
Bevacizumab (Avastin®), a humanized variant of an anti-VEGF neutralizing monoclonal antibody, is the first antiangiogenic agent to be approved by the FDA for the treatment of cancer.5, 7 Bevacizumab was approved for the treatment of metastatic colorectal cancer8 and non-small cell lung cancer9 in combination with chemotherapy. The drug is presently being tested in several phase III studies in combination with chemotherapy. While bevacizumab is generally well tolerated, some significant toxicities were infrequently observed, including hypertension, gastrointestinal perforation and arterial thromboembolic complications (reviewed by Ferrara et al5).
In addition to agents blocking VEGF itself, a variety of small molecule receptor tyrosine kinase (RTK) inhibitors targeting the VEGF receptors including SU11248 (Sutent®) and Bay 43–9006 (sorafenib) have been developed. Sorafenib was initially identified as a raf kinase inhibitor and was later shown to inhibit several RTKs including VEGFRs. An interim phase III analysis indicates that Sorafenib monotherapy results in a significant increase in progression-free survival in patients with advanced renal cell carcinoma. SU11248 inhibits VEGFRs, PDGFR, c-kit and Flt-3 and has efficacy in imatinib-resistant gastrointestinal stromal tumor.10 Other anti-VEGF agents including VEGF-Trap (Regeneron), a soluble receptor targeting VEGF, VEGF-B and PlGF; an antisense oligonucleotide VEGF-AS (Vasgene Therapeutics Inc.) targeting VEGF, VEGF-C and VEGF-D are at various stages of clinical development.11 Overall, characterization of VEGF signaling pathway led to the identification of several target molecules with promising therapeutic potentials.
Anti-VEGF treatments have also applications in disorders other than cancer. Wet (neovascular) AMD is the most common cause of severe, irreversible vision loss in the elderly.12 Several pharmacologic agents have been approved by the FDA for the treatment of neovascular AMD. One is verteporfin (Visudyne®) photodynamic therapy (PDT).13 The other is Pegaptanib sodium (Macugen®) approved in December 2004 for all angiographic subtypes of neovascular AMD.14 Although both treatments can slow the progression of vision loss, only a small percentage of treated patients experience any improvement in visual acuity. The third FDA approved treatment for wet AMD is ranibizumab (Lucentis™), a recombinant, humanized Fab that binds to and neutralizes the biological activities of all human VEGF-A isoforms.15 Ranibizumab has been evaluated in two large, phase III, multicenter, pivotal trials in different neovascular AMD patient populations. The MARINA trial randomized subjects with minimally classic (less than 50% of the lesion consisting of classic CNV) or occult without classic CNV to monthly sham injections or monthly intravitreal injections of one of two doses of ranibizumab.16 On average, ranibizumab-treated subjects gained vision at 1 year compared with baseline while sham-injection subjects lost vision. A significantly larger percentage of subjects treated with ranibizumab gained ≥15 letters at 1 year than did the sham-injection group. The visual acuity benefits observed at 1 year were maintained through the second year.16 The ANCHOR trial randomized subjects with predominantly classic CNV to verteporfin PDT.17 On average, ranibizumab-treated subjects gained vision at 1 year compared with baseline, while verterporfin PDT subjects lost vision, and a significantly larger percentage of subjects treated with ranibizumab gained ≥15 letters at 1 year than did the verteporfin PDT group.
Resistance to Anti-VEGF treatment
Many cancer patients treated with VEGF inhibitors survive longer, but they eventually die due to resistance to the treatment. Potentially, other angiogenic pathways may replace VEGF as the disease progresses. Alternatively, tumor cell variants that are ‘hypoxic resistant’ and thus less dependent on angiogenesis are selected and outgrow the sensitive tumor cells.18 Moreover, remodeling of tumor vessels that results in the generation of mature, stabilized vessels that are less responsive to angiogenesis inhibitors is considered to be one of the mechanisms of resistance. In addition to tumor cells, resistance may originate from the nontumor cell compartment known as stromal, even though it is less prone to genetic instability and mutations.3 Tumor stroma comprises a variety of cell types such as fibroblasts and bone marrow-derived cells (BMDCs) and may be a source of various proangiogenic factors. Therefore, investigating stromal–tumor cells interactions is of particular interest in understanding resistance to antiangiogenic treatments.
Tumor associated fibroblasts (TAFs) are one of the major components of stromal cells in many caner types. Recently, TAFs were reported to be a main source of stromal-derived factor 1 (SDF-1), leading to the recruitment of endothelial progenitor cells (EPCs) or myeloid cells in the tumor microenvironment.19 BMDCs that infiltrate the tumor microenvironment in several tumor types might also provide a mechanism of escape from antiangiogenic therapy. BMDCs have been proposed to participate in tumor angiogenesis through at least two different mechanisms, (i) direct incorporation in the tumor vasculature and (ii) as a source of proangiogenic factors. Early studies by Lyden et al,20 suggested a direct role for endothelial progenitor cells (EPCs), originated from BMDCs, in the tumor vasculature. In contrast, using a genetic model of an endothelial-specific inducible gene, Gothert et al,21 observed a lack of participation of BMD-EPCs in the tumor vasculature.
Recent studies have identified a population of Tie2 expressing monocyte (TEM) that promotes tumor growth through the secretion of angiogenic factors.22 Also, various tumor-infiltrating hematopoietic cells such as T- and B-lymphocytes, monocytes/macrophages have also been suggested to secrete angiogenic factors. Much research focused on CD11b+Gr1+ cells that include cells of monocyte and granulocytic lineages.23 Recent studies indicate that neutrophils (a subpopulation of CD11b+Gr1+ cells) are a source of matrix metalloproteinase (MMP)-9 and play a role in the induction of angiogenic switch in a transgenic model of β-cell carcinogenesis.24
Figure 1 illustrates a model to explain the infiltration of BMDCs in several tumor types. Secretion of cytokines/chemokines by tumor cells or TAFs such as SDF-1, VEGF, granulocyte macrophage colony stimulating factor (GM-CSF) and G-CSF appears to be a key step to trigger BMDCs and results in their mobilization (step 1), transendothelial migration (step 2) and homing (step 3) to the tumors. An amplification process may result in an environment in which angiogenic factors secreted by stromal cells and/or direct contribution of BMDCs to tumor vasculature result in the development of resistance to the antiangiogenic treatment.

Potential mechanisms of recruitment of BMDCs in tumors. Tumors cells appear to initiate the recruitment of stromal cells through secretion of several angiogenic factors and cytokines. In addition, secretion of variety of cytokines/chemokines by tumor cells and tumor associated fibroblasts (TAFs), including SDF-1, GM-CSF, VEGF and G-CSF results in the mobilization (1), transendothelial migration (2) and homing (3) of BMDCs to the tumors. Subsequently, secretion of chemokines/cytokines and angiogenic factors by BMDCs further amplify the migration of several hematopoieitc lineages to the tumors.
Finally, tumor-associated endothelial cells (TAECs) appear to be one more possible source of resistance to inhibitors of angiogenesis. Recent studies suggest that in some cases TAECs may not represent a genetically stable, nontransformed, compartment as previously thought but show various cytogenetic abnormalities.25
Conclusion and future prospects
There is now compelling evidence that targeting angiogenesis in general and VEGF signaling in particular is a meaningful approach for the therapy not only of cancer but also of wet AMD.
A future challenge is establishing optimal dosages and therapeutic regimens. It appears likely that cancer therapy will be in most cases combinatorial. Antiangiogenic agents therefore, will need to be combined with cytotoxic chemotherapy and/or targeted therapies. Indeed, preclinical studies have demonstrated additive or synergistic effects between angiogenesis inhibitors and a variety of anticancer agents (reviewed by Ferrara and Kerbel6). There is considerable debate regarding the mechanisms of potentiation with chemotherapy. In fact it seems counterintuitive that agents, which reduce tumor blood flow such as angiogenesis inhibitors may enhance the efficacy of cytotoxic agents. One hypothesis postulates that antiangiogenic agents may, in some circumstances, ‘normalize’ the tumor vasculature, resulting in improved delivery of chemotherapy into tumor cells.26 Also, it has been proposed that administration of low-dose chemotherapy at close regular intervals (‘metronomic therapy’) preferentially damages tumor vessels such that the combination with antiangiogenic agents (eg VEGF blockers) amplifies the antivascular effects, leading to enhanced killing of tumor cells.27
Future investigations are also required to identify cellular and molecular mechanisms underlying resistance to anti-VEGF treatment, which may provide additional therapeutic targets. Given the important role of the stromal cell compartment in promoting tumor growth, chemokines/cytokines that are able to recruit BMDCs to the tumors and promote proliferation of endothelial cells seem to be attractive candidates for future antiangiogenic treatments.
References
Coultas L, Chawengsaksophak K, Rossant J . Endothelial cells and VEGF in vascular development. Nature 2005;438:937–945.
Ferris III FL, Fine SL, Hyman L . Age-related macular degeneration and blindness due to neovascular maculopathy. Arch Ophthalmol 1984;102:1640–1642.
Ferrara N, Gerber HP, LeCouter J . The biology of VEGF and its receptors. Nat Med 2003;9:669–676.
Hicklin DJ, Ellis LM . Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J Clin Oncol 2005;23:1011–1027.
Ferrara N, Hillan KJ, Gerber HP, et al Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov 2004;3:391–400.
Ferrara N, Kerbel RS . Angiogenesis as a therapeutic target. Nature 2005;438:967–974.
Kim KJ, Li B, Winer J, et al. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature 1993;362:841–844.
Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 2004;350:2335–2342.
Sandler A, Gray R, Perry MC, et al Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell-lung cancer. N Engl J Med 2006;355:2542–2550.
Smith JK, Mamoon NM, Duhe RJ . Emerging roles of targeted small molecule protein-tyrosine kinase inhibitors in cancer therapy. Oncol Res 2004;14:175–225.
Jain RK, Duda DG, Clark JW, et al. Lessons from phase III clinical trials on anti-VEGF therapy for cancer. Nat Clin Pract Oncol 2006;3:24–40.
Congdon N, O'Colmain B, Klaver CC, et al. Causes and prevalence of visual impairment among adults in the United States. Arch Ophthalmol 2004;122:477–485.
Schmidt-Erfurth U, Miller JW, Sickenberg M, et al. Photodynamic therapy with verteporfin for choroidal neovascularization caused by age-related macular degeneration: results of retreatments in a phase 1 and 2 study. Arch Ophthalmol 1999;117:1177–1187.
Gragoudas ES, Adamis AP, Cunningham Jr ET, et al. Pegaptanib for neovascular age-related macular degeneration. N Engl J Med 2004;351:2805–2816.
Ferrara N, Damico L, Shams N, et al. Development of ranibizumab, an anti-vascular endothelial growth factor antigen binding fragment, as therapy for neovascular age-related macular degeneration. Retina 2006;26:859–870.
Rosenfeld PJ, Brown DM, Heier JS, et al. Ranibizumab for neovascular age-related macular degeneration. N Engl J Med 2006;355:1419–1431.
Brown DM, Kaiser PK, Michels M, et al. Ranibizumab versus verteporfin for neovascular age-related macular degeneration. N Engl J Med 2006;355:1432–1444.
Yu JL, Coomber BL, Kerbel RS . A paradigm for therapy-induced microenvironmental changes in solid tumors leading to drug resistance. Differentiation 2002;70:599–609.
Orimo A, Gupta PB, Sgroi DC, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005;121:335–348.
Lyden D, Hattori K, Dias S, et al. Impaired recruitment of bone-marrow-derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nat Med 2001;7:1194–1201.
Gothert JR, Gustin SE, van Eekelen JA, et al. Genetically tagging endothelial cells in vivo: bone marrow-derived cells do not contribute to tumor endothelium. Blood 2004;104:1769–1777.
De Palma M, Venneri MA, Galli R, et al. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell 2005;8:211–226.
Yang L, DeBusk LM, Fukuda K, et al. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell 2004;6:409–421.
Nozawa H, Chiu C, Hanahan D . Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proc Natl Acad Sci USA 2006;103:12493–12498.
Hida K, Hida Y, Amin DN, et al. Tumor-associated endothelial cells with cytogenetic abnormalities. Cancer Res 2004;64:8249–8255.
Jain RK . Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science 2005;307:58–62.
Kerbel RS . Antiangiogenic therapy: a universal chemosensitization strategy for cancer? Science 2006;312:1171–1175.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Shojaei, F., Ferrara, N. Antiangiogenesis to treat cancer and intraocular neovascular disorders. Lab Invest 87, 227–230 (2007). https://doi.org/10.1038/labinvest.3700526
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.3700526
Keywords
This article is cited by
-
Targeting soluble CD146 with a neutralizing antibody inhibits vascularization, growth and survival of CD146-positive tumors
Oncogene (2016)
-
Functional characterization of a VEGF-A-targeting Anticalin, prototype of a novel therapeutic human protein class
Angiogenesis (2016)
-
Anti-vascular endothelial growth factor antibody attenuates inflammation and decreases mortality in an experimental model of severe sepsis
Critical Care (2013)
-
The subconjunctival use of cetuximab and bevacizumab in inhibition of corneal angiogenesis
Graefe's Archive for Clinical and Experimental Ophthalmology (2012)
-
SR16388: a steroidal antiangiogenic agent with potent inhibitory effect on tumor growth in vivo
Angiogenesis (2011)
