
Abstract
Despite extensive investigation, the cellular mechanisms responsible for neuroleptic actions remain elusive. We have previously shown that neuroleptics modulated the expression of some members of the ligand-activated transcription factors (nuclear receptors) including the nerve-growth factor inducible gene B (NGFI-B or Nur77) and retinoid X receptor (RXR) isoforms. Using genetic and pharmacological approaches, we investigated the role of NGFI-B and retinoids in acute behavioral and biochemical responses to dopamine antagonists. NGFI-B knockout (KO) mice display a profound alteration of haloperidol-induced catalepsy and striatal neuropeptide gene expression. Haloperidol-induced increase of striatal enkephalin mRNA is totally abolished in NGFI-B KO mice whereas the increase of neurotensin mRNA expression is reduced by 50%. Interestingly, catalepsy induced by raclopride, a specific dopamine D2/D3 antagonist is completely abolished in NGFI-B-deficient mice whereas the cataleptic response to SCH 23390, a dopamine D1 agonist, is preserved. Accordingly, the effects of haloperidol on striatal c-fos, Nor-1, and dynorphin mRNA expression are also preserved in NGFI-B-deficient mice. The cataleptic response and the increase of enkephalin mRNA expression induced by haloperidol can also be suppressed by administration of retinoid ligands 9-cis retinoic acid and docosahexaenoic acid. In addition, we demonstrate that haloperidol enhances colocalization of NGFI-B and RXRγ1 isoform mRNAs, suggesting that both NGFI-B and a RXR isoform are highly coexpressed after haloperidol administration. Our data demonstrate, for the first time, that NGFI-B and retinoids are actively involved in the molecular cascade induced by neuroleptic drugs.
Similar content being viewed by others

INTRODUCTION
Antipsychotic drugs or neuroleptics are used to reduce symptoms of schizophrenia. Although the putative mechanism of action of neuroleptics has been extensively investigated, the underlying cellular events responsible for their clinical efficacy, as well as for their undesired side effects remain elusive. Neuroleptics are divided into two classes. Typical or conventional neuroleptics (such as haloperidol) are defined as drugs that improve positive symptoms of schizophrenia (eg hallucinations), but have a high propensity to cause a variety of extrapyramidal symptoms (EPS). EPS are among the most frequent problems experienced with conventional antipsychotic medication. It is estimated that as many as 90% of patients treated with standard neuroleptics develop EPS (Kane, 2001). Atypical or new generations of neuroleptics (such as clozapine or olanzapine) improve both positive and negative symptoms of schizophrenia (eg low affect) with lower propensity to induce motor side effects (Meltzer, 1995). However, serious and unexpected new side effects, including agranulocytosis (clozapine) and dramatic weight gains, have emerged with the used of the new generations of neuroleptics (Allison and Casey, 2001). Owing to these side effects and also for ecomonic considerations, haloperidol, a conventional neuroleptic, is still commonly prescribed. Blockade of the dopamine D2 receptor in the ventral striatum is thought to underlie some of the antipsychotic effect of neuroleptics (Seeman, 1995). However, interaction with the dopamine D2 receptor in the dorsal striatum is also responsible for EPS induced by these drugs (Seeman, 1995). Although the interaction of neuroleptics with neurotransmitter receptors is well characterized, intracellular signaling pathways triggered by this interaction remain mostly unexplored. However, the ability of these drugs to induce the expression of transcription factors and neuropeptides has led to the suggestion that changes in gene expression might be responsible for certain antipsychotic drug actions (Hiroi and Graybiel, 1996; Steiner and Gerfen, 1998).
Nuclear receptors represent an important family of proteins regulating gene expression. Several lines of evidence from our laboratory suggest the possible involvement of nuclear receptor family of transcription factors in the effects of antipsychotic drugs. We have shown that typical and atypical antipsychotics induced contrasting patterns of expression of nerve growth factor-inducible gene B (NGFI-B, also known as NR4A1 or Nur77) an orphan nuclear receptor closely related to members of the steroid/thyroid hormone receptor family (Hazel et al, 1988; Milbrandt, 1988), after acute and chronic administration (Beaudry et al, 2000). We have also shown that acute and chronic neuroleptic treatment modulate the expression of other transcription factors belonging to the nuclear receptor family including retinoic acid receptors (RARs) and retinoid X receptors (RXRs) (Langlois et al, 2001).
Retinoic acids, through interaction with ligand-activated transcription factors RAR and RXR, regulate the expression of numerous target genes and are particularly active during brain development (Chambon, 1996; Maden, 2002). RARs are specifically involved in retinoid signaling whereas RXRs also participate in many other signaling events by serving as heterodimerization partners not only for RARs, but also for the vitamin D receptor, the thyroid hormone receptors (TR) and different orphan members of the nuclear receptor family of transcription factors such as Nurr1 and NGFI-B (Nur77) (Perlmann and Jansson, 1995). Several lines of evidence strongly suggest that in addition to have a key role during development, retinoic acid might have an important role in dopamine-innervated basal ganglia in the mature brain. Both RARβ and RXRγ isoforms are expressed in the striatum, nucleus accumbens and olfactory tubercle of both newborn and adult rats (Krezel et al, 1999; Saga et al, 1999; Zetterström et al, 1999). It has been shown that RARβ- and RXRγ-deficient mice demonstrate impaired locomotion, dopamine signaling (Krezel et al, 1999) and an altered response to dopamine antagonists (Saga et al, 1999).
The aim of the present study was to assess the involvement of NGFI-B and retinoids on the biochemical and behavioral effects of neuroleptics. We studied and compared the effects of acute neuroleptic administration in wild-type (WT) mice and in a NGFI-B KO strain of mice (Lee et al, 1995). We also investigated the effects of retinoid ligands on biochemical and behavioral responses induced by a neuroleptic.
EXPERIMENTAL PROCEDURES
Animal Care and Treatments
All procedures, including means to minimize discomfort, were reviewed and approved by the Laval University Animal Care Committee. NGFI-B KO mice were developed by the group of Dr Milbrandt at the University of Washington (St Louis, Missouri, USA) (Lee et al, 1995). They are healthy and reproduce normally (Crawford et al, 1995; Lee et al, 1995). They were produced in a mixed background and have been backcrossed into the C57BL/6 strain for at least 10 generations to reduce background heterogeneity (Jeff Milbrandt, personal communication). NGFI-B-deficient and WT (C57BL/6) mice (Charles River, Canada, weighing 20–25 g) were acutely treated with the different dopamine receptor antagonists (0.25 ml, i.p.) at various doses (haloperidol 0.1, 0.5, and 1 mg/kg; raclopride 1.25 mg/kg and SCH 23390 0.75 mg/kg). Saline was used as vehicle for haloperidol, raclopride, and SCH 23390 (RBI, Oakville, ON, Canada). The animals were killed by decapitation under CO2 anesthesia. For the evaluation of neuropeptide (preproenkephalin, ENK; prodynorphin, DYN, and neurotensin/neuromedin N precursor, NT) mRNA levels, animals were killed 5 h after drug injection. For the evaluation of immediate-early gene expression (NGFI-B, Nor-1, and c-fos), the animals were killed 1 h after drug administration (catalepsy was not measured in those animals). The vitamin A derivative, 9-cis retinoic acid (9-cis RA) and the polyunsaturated fatty acid (PUFA) docosahexaenoic acid (DHA) were administered as stable suspensions in 4% ethanol and 8% PEG-600 in sterile water (pH adjusted between 5.5 and 6.5). In the experiments involving combined treatments with retinoid ligands, the group of animals treated with haloperidol alone also received the 9-cis RA and DHA vehicle (4% ethanol and 8% PEG-600 in sterile water). After decapitation, brains were rapidly removed and immediately immersed into cold isopentane (−40°C) for a few seconds and kept at −80°C until used.
Catalepsy
Catalepsy was evaluated using the inclined plan procedure (Dobner et al, 2001). Mice were placed in a mesh wire grid inclined to an angle of 70°. The catalepsy time was defined as the time for the mice to move all four paws. The test was performed for a maximal duration of 180 s. Catalepsy was measured at 15, 30, 60, 90, and 120 min following dopamine receptor antagonist administration (without or with retinoid ligands). Average catalepsy times represent the mean catalepsy time obtained at 60 and 90 min after injection.
Autoradiography
For determination of the density of dopamine D2 receptor-binding sites, a buffer containing 50 mM Tris-HCl, 120 mM NaCl, 5 mM KCl, 1.5 mM CaCl2, 4 mM MgCl2, and 1 mM EDTA (pH 7.4) was used with 3 nM [3H]raclopride (specific activity: 79.3 Ci/mmol, Dupont NEN™, Guelph, ON, Canada) (Tremblay et al, 1999). A measure of 1 M (+)-butaclamol (RBI, Natick, MA) was used to determine nonspecific binding. Slides were exposed against tritium sensitive films ([3H]hyperfilms, Amersham, Oakville, ON, Canada) for 2 weeks. Quantification of autoradiograms was performed as previously described (Tremblay et al, 1999).
For determination of the density of dopamine D1 receptor-binding sites, rat brain sections were preincubated in a buffer containing 15 mM Tris-HCl, 120 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 0.1% ascorbic acid, and 0.1 mM EDTA (pH 7.4) for 15 min at room temperature and then incubated in the same buffer containing 0.2 nM [N-methyl-3H]SCH 23390 (specific activity: 85 Ci/mmol, Amersham, Oakville, ON, Canada) for 1 h at room temperature. A measure of 1 M SCH 23390 (RBI, Natick, MA) was used to determine nonspecific binding. Slides were exposed against BiomaxMR sensitive films (Kodak, New Haven, CT) for 2 weeks.
Single and Double In Situ Hybridization Procedures
Cryostat coronal brain sections (12 μm) were mounted onto Snowcoat X-tra™ slides (Surgipath, Winnipeg, MA, Canada) and stored at −80°C until used. Brain sections were fixed in 4% paraformaldehyde at 4°C for 20 min. For single in situ hybridization, specific [35S]UTP-radiolabeled complementary RNA (cRNA) probes were used. The NGFI-B, c-fos, ENK, DYN, and NT probe preparations have been described in detail elsewhere (Tremblay et al, 1999; Beaudry et al, 2000; Langlois et al, 2001). The mouse Nor-1 probe was generated from a PCR fragment of 393 bp (from nucleotide 572 to 964) (Maltais and Labelle, 2000) subcloned into pBluscript SK+ linearized with HindIII to generate the antisense cRNA. The radiolabeled probe was generated as previously described (Langlois et al, 2001). In situ hybridization of the riboprobes with tissue sections were done at 55–58°C, overnight, in a standard hybridization buffer containing 50% formamide (Beaudry et al, 2000; Langlois et al, 2001). Tissue sections were then apposed against BiomaxMR (Kodak, New Haven, CT) radioactive sensitive films for 2–10 days. Quantification of autoradiograms was performed using computerized analysis (NIH Image software, Wayne Rasband, NIMH). Optical gray densities were transformed into nCi/g tissue equivalent using standard curves generated with 14C-microscales (Amersham, Oakville, ON, Canada). Brain areas investigated included the dorsolateral (StDL) and dorsomedial (StDM) portion of the striatum, the shell (AcSh) and core (AcC) of the nucleus accumbens, medial prefrontal (mPFC) and cingulate (CC) cortices.
The double in situ hybridization procedure was performed as previously described (Beaudry et al, 2000). Briefly, the proportion of colocalization of the NGFI-B transcript with the RXRγ1 mRNA in vehicle- and APD-treated animals was evaluated using simultaneous double in situ hybridization with a [35S]UTP-labeled NGFI-B probe and a nonradioactive digoxygenin (Dig)-labeled RXRγ1 probe. The RXRγ1 cRNA probe was labeled with Dig using the Riboprobe System of Promega (Madison, MA) with the Dig-RNA labeling mix (Roche Diagnostics, Laval, Qc, Canada). Double in situ hybridization was performed in the same conditions as for single in situ procedure. The dig-cRNA probe (about 10 ng) was simply added in the same hybridization solution with the radioactive (4 × 106 cpm) cRNA probe for NGFI-B. An additional step using a 50% formamide solution in 2 × SSC buffer after hybridization was performed to reduce nonspecific Dig labeling. Revelation of the Dig-labeling was performed with an anti-Dig antibody conjugated to alkaline phosphatase (Boehringer Mannheim, Laval, Qc, Canada) and evidenced using a nitroblue tetrazolium chloride, 5-bromo-4-chloro-3-indolyl phosphate and levamisole chromogen solution (Beaudry et al, 2000). Slides were then dipped in LM-1 photographic emulsion (Amersham, Oakville, ON, Canada) melted at 43°C, air-dried and stored in the dark for 12 days at 4°C. The emulsion was developed in D-19 developer and fixed (Kodak, New Haven, CT). Slides were coverslipped using a water-soluble mounting medium (Permafluor, Lipshaw Immunon, Pittsburgh, PA). Single- or double-labeled cells were visualized and manually counted under bright-field illumination with a Zeiss photo microscope at a magnification of × 400. Neuron counting was performed on four different sections obtained from a total of three animals per group investigated. Fields for quantification were taken within the StDL region.
Statistical Analysis
Data were compared using an analysis of variance (one-way ANOVA), followed by a Fisher probability of least significant difference (PLSD) test.
RESULTS
Neuroleptic-Induced Catalepsy is Strongly Reduced in NGFI-B-Deficient Mice
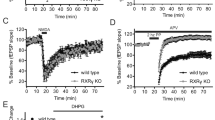
Haloperidol induces a strong dose-dependent cataleptic response in WT (C57BL/6) mice, but this cataleptic response is dramatically reduced in the NGFI-B (−/−) mice (Figure 1a–c). In addition, the cataleptic response induced by raclopride, a specific D2/D3 receptor antagonist, is completely blocked in the NGFI-B-deficient mice (Figure 1d), whereas the catalepsy induced by SCH 23390, a selective D1-like receptor antagonist, is not affected (Figure 1e). The reduced effect of D2 receptor antagonists (haloperidol and raclopride) is not due to a lower expression of the D2 receptor in the KO mice, since the levels of dopamine D2 receptor-binding sites measured with [3H]raclopride binding are normal in the nucleus accumbens and the StDM, but are significantly upregulated in the StDL of NGFI-B-deficient mice (Figure 2). Striatal levels of the dopamine D1-binding site, measured with the specific D1/D5 receptor ligand [3H]SCH 23390, are not significantly changed in the NGFI-B KO mice compared to WT mice (StDL: WT, 187±6 and KO, 170±9 fmol/mg of protein; StDM: WT, 149±5 and KO, 132±7 fmol/mg of protein, p=0.06).

Catalepsy induced by dopamine D2 receptor antagonists is strongly reduced in NGFI-B KO mice. (a–c) Time course of haloperidol (HAL)-induced catalepsy (HAL: (a), 0.1 mg/kg; (b), 0.5 mg/kg; (c), 1 mg/kg) in NGFI-B KO (NGFI-B (−/−)) and WT (NGFI-B (+/+)) mice. At all doses, NGFI-B-deficient mice show a significant reduction in the cataleptic response compared to WT mice (*p<0.05, **p<0.01 and ***p<0.001 vs NGFI-B (+/+) HAL-treated mice and ##p<0.01 vs vehicle (VEH)-treated NGFI-B (+/+) mice). Each time point represents the mean±SEM from 8–10 animals per group. (d) Effect of raclopride (1.25 mg/kg, i.p.), a specific dopamine D2/D3 receptor antagonist, on the cataleptic behavior in WT and NGFI-B KO mice. Raclopride-induced catalepsy is completely abolished in NGFI-B (−/−) mice. Each time point represents the mean±SEM from five to seven animals per group (**p<0.01 and ***p<0.001 vs NGFI-B (+/+) group). (e) WT and NGFI-B (−/−) mice display a similar cataleptic response to SCH 23390 (0.75 mg/kg, i.p.), a dopamine D1 receptor antagonist.

NGFI-B-deficient mice displayed higher levels of dopamine D2 receptors in the StDL compared to WT mice. D2 receptor levels were unchanged in other striatal areas; StDM and AcSh. Levels of dopamine D2/D3-binding sites were measured with [3H]raclopride (3 nM) autoradiography. Each histogram bar represents the mean±SEM from five to seven animals per group (**p<0.01 vs NGFI-B (+/+) mice).
Specific Effects of Haloperidol on Gene Expression are Disrupted in NGFI-B (−/−) Mice
Acute haloperidol administration increases both ENK (Figure 3a, b) and neurotensin/neuromedin N precursor (NT) (Figure 3c, d) mRNA levels in the StDL of WT mice. Haloperidol-induced upregulation of ENK mRNA is totally abolished and NT mRNA increase is reduced by 50% in NGFI-B-deficient mice in the StDL (Figure 3b, d). Similar effects are observed in other striatal areas (Table 1). The effect of haloperidol on NT mRNA levels is completely abolished in the AcSh in NGFI-B-deficient mice (Table 1).

The effects of haloperidol on striatal neuropeptide expression associated to dopamine D2 receptors are reduced in NGFI-B KO mice. Effects of HAL (1 mg/kg) on the expression of the preproenkephalin (ENK) mRNA (a and b) and neurotensin/neuromedin N precursor (NT) (c and d) in NGFI-B-deficient (NGFI-B(−/−)) and WT (NGFI-B(+/+)) mice using in situ hybridization. Representative autoradiograms generated with a specific [35S]UTP-labeled ENK and NT riboprobes are shown in panels (a) and (c), respectively. Histograms of the effects of HAL in the StDL are shown in panels (b) and (d). HAL administration increased ENK (b) and NT (d) mRNA levels in the StDL in NGFI-B (+/+) mice. In NGFI-B (−/−) mice, the acute effect of haloperidol on the striatal level of ENK mRNA is abolished and its effect on NT mRNA is reduced by 50%. Values of mRNA levels are expressed in nCi/g of tissue equivalent. Each histogram bar represents the mean±SEM from 8–10 animals per group (**p<0.01 and ***p<0.001 vs respective VEH groups and ##p<0.01 vs HAL-treated NGFI-B (+/+) mice).
On the contrary, the effects of haloperidol on the expression of DYN (Figure 4a, b), c-fos (Figure 4c, d), and Nor-1 (Figure 4e, f), a close homologue of NGFI-B (Paulsen et al, 1995), are similar in WT and NGFI-B (−/−) mice. However, the effect of haloperidol on Nor-1 mRNA levels is significantly higher in NGFI-B-deficient mice compared to WT mice (Figure 4f). Basal levels of striatal neuropeptide transcripts and immediate-early genes (c-fos and Nor-1) in NGFI-B (−/−) mice are equivalent to those of the untreated WT mice (Figures 3 and 4). Similar effects are observed in other striatal areas and other brain areas investigated (Table 1).

NGFI-B-deficient mice displayed similar modulation of DYN, c-fos, and Nor-1 mRNA levels compared to WT mice after haloperidol administration. (a, c, e) Representative examples of autoradiograms generated with the specific [35S]UTP-labeled riboprobe after in situ hybridization of the DYN (a), c-fos (c), and Nor-1 (e) mRNAs in the striatum of VEH- and HAL-treated mice. (b, d, f) Quantification of the effects of haloperidol in the StDL. HAL administration reduces DYN mRNA levels (b) in the StDL in both NGFI-B (+/+) and (−/−) mice, whereas it increases c-fos (d) with equivalent intensity in both strains. The effects of HAL on Nor-1 (f) mRNA levels are significantly higher in NGFI-B-deficient mice compared to WT mice (#p<0.05). Each histogram bar represents the mean±SEM from 8–10 animals per group (*p<0.05, **p<0.01 and ***p<0.001 vs respective VEH-treated group).
NGFI-B and RXRγ1 are Highly Colocalized after Haloperidol Treatment
The StDL, which is associated with the control of locomotor behaviors, displays a very high RXRγ1 mRNA level (Figure 5a). Interestingly, the distribution of the RXRγ1 mRNA in the striatum corresponds to the localization of the NGFI-B mRNA after the administration of haloperidol (Figure 5a). Acute administration of haloperidol, as used herein, did not significantly change RXRγ1 mRNA levels in the striatum (Figure 5b). The NGFI-B gene KO did not modify the basal expression of this retinoid receptor isoform (Figure 5b). However, double in situ hybridization experiments indicate that haloperidol induces a dramatic increase in the percentage of colocalization of NGFI-B and RXRγ1 mRNAs in the StDL (Figure 5c, d). This suggests that both transcription factors are highly coexpressed upon haloperidol administration. Therefore, we hypothesized that retinoid ligands may interfere with neuroleptic-induced catalepsy and gene expression.

Haloperidol administration increases the colocalization of NGFI-B and RXRγ1 transcripts. (a) Comparison of the distribution of NGFI-B and RXRγ1 mRNAs in VEH- and HAL-treated (1 mg/kg) mice in the caudal striatum. Acute administration of haloperidol strongly increases NGFI-B mRNA levels in a striatal area expressing a high density of the RXRγ1 mRNA in basal conditions. (b) Effects of HAL (1 mg/kg) on RXRγ1 mRNA levels in the StDL in NGFI-B (+/+) and (−/−) mice. Each histogram bar represents the mean±SEM from five to seven animals per group. (c) Representative image of the colocalization of NGFI-B and RXRγ1 mRNAs in the StDL after acute haloperidol administration. The double in situ hybridization procedure was performed with a [35S]UTP-labeled NGFI-B probe (silver grains) combined with a digoxigenin-labeled RXRγ1 probe (dark staining revealed with an anti-Dig antibody conjugated to alkaline phosphatase) (Beaudry et al, 2000). Arrowheads indicate positive NGFI-B-labeled cells (silver grains); thin arrows show positive RXRγ1 cells (dark depots) and bold arrows represent cells positive for both NGFI-B and RXRγ1 transcripts. (d) Acute HAL administration strongly increase the percentage of colocalization of NGFI-B and RXRγ1 mRNAs in the StDL in WT animals (***p<0.001 vs VEH-treated animals). Values are expressed in percentage of colocalization (double-labeled cells) compared to VEH-treated animals and represent the mean±SEM from three animals.
Retinoid Ligands can Suppress Haloperidol-Induced Catalepsy
To test this hypothesis, we administered 9-cis RA and DHA, two potent RXR ligands, in combination with haloperidol to WT mice (Figure 6). It has been previously shown that 9-cis RA strongly enhances the transcriptional activity of the NGFI-B/RXR heterodimer in vitro (Heyman et al, 1992; Vivat et al, 1997). More recently, DHA has been identified as a natural ligand for retinoid receptors (de Urquiza et al, 2000; Egea et al, 2002). Administration of 9-cis RA in combination with haloperidol dose-dependently reduces the severity of drug-induced catalepsy (Figure 6a, b), whereas it has no behavioral effect when injected alone (Figure 6a). A similar dose–response effect is observed when increasing concentrations of DHA are injected in combination with haloperidol (0.5 mg/kg) treatment (Figure 6c, d). Interestingly, the highest dose of DHA (100 mg/kg) completely suppresses haloperidol-induced catalepsy (Figure 6d). Note also that the effects of retinoid ligands become apparent only after 90 min after injections of the drugs, while haloperidol starts to significantly induce catalepsy at 30 min (Figure 6a, c).

Coadministration of retinoid ligands strongly reduces haloperidol-induced catalepsy. (a) Representative example of the time course of catalepsy induced by HAL (0.5 mg/kg) alone or in combination with 9-cis retinoic acid (9-cis RA, 5 mg/kg). Administration of 9-cis RA alone remains without effect. Each time point represents the mean±SEM from six to eight animals (*p<0.05 and **p<0.01 vs corresponding HAL alone time point). (b) Administration of 9-cis RA (2.5, 5, and 10 mg/kg) dose-dependently reduces the average catalepsy time induced by a single dose of haloperidol (0.1 or 0.5 mg/kg). Each histogram bar represents the mean±SEM from six to eight animals per group (*p<0.05 vs respective HAL alone group). (c) Representative example of the time course of catalepsy induced by HAL (0.5 mg/kg) alone or in combination with DHA (50 mg/kg). Administration of DHA alone remains without effect. Each time point represents the mean±SEM from six to eight animals (**p<0.01 vs corresponding HAL alone time point). (d) Administration of DHA (20, 50, and 100 mg/kg) also dose-dependently reduces HAL (0.5 mg/kg)-induced catalepsy. Each histogram bar represents the mean±SEM from six to eight animals per group (*p<0.05 vs respective HAL alone group).
Retinoid Ligands Prevent Haloperidol-Induced Enkephalin mRNA Upregulation
Administration of 9-cis RA (10 mg/kg) or DHA (100 mg/kg) in combination with haloperidol (0.5 mg/kg) prevents or abolishes the haloperidol-induced ENK gene expression in the striatum of WT mice (Figure 7). Administration of 9-cis RA in combination with haloperidol significantly reduced below controls ENK mRNA levels in the nucleus accumbens, whereas acute administration of retinoid ligands alone had no effect on ENK expression (9-cis RA alone in StDL: 86.2±12.0, not significant). Note that the 0.5 mg/kg dosage of haloperidol used here gave a similar increase (about 50%) of the ENK mRNA level as the dosage of 1 mg/kg used in Figure 3.

Effects of retinoid ligands on haloperidol-induced enkephalin (ENK) expression. An acute injection of HAL (0.5 mg/kg) significantly increases ENK mRNA levels in the StDL and the AcSh, whereas ENK mRNA levels are not significantly upregulated in the StDM and the AcC. Coadministration of the same dose of HAL in combination with 9-cis retinoic acid (9-cis RA, 10 mg/kg) or DHA (100 mg/kg) completely suppresses or prevents HAL-induced ENK mRNA upregulation. ENK mRNA levels are even reduced below VEH group levels in the nucleus accumbens (AcSh and AcC) with combination of HAL and 9-cis RA. Values are expressed in percentage (%) from the control group (VEH) and represent the mean±SEM from six to eight animals per group (*p<0.05 and **p<0.01 vs respective VEH group).
DISCUSSION
The present results establish a role for NGFI-B in the molecular cascade induced by dopamine D2 receptor antagonist leading to catalepsy and specific modulations of neuropeptide gene expression. In addition, we show that the retinoid ligands 9-cis RA (a vitamin A derivative) and DHA (an ω-3 PUFA) can strongly reduce or even suppress haloperidol-induced catalepsy and enkephalin opioid gene expression, suggesting that retinoids might also be involved in this molecular cascade. The high percentage of colocalization of both NGFI-B and RXRγ1 transcripts after haloperidol treatment in the StDL, a brain region involved in the control of locomotor behaviors, suggest that these effects may take place in the same striatal cell population.
Haloperidol-induced catalepsy is thought to reproduce acute EPS observed in humans (Hoffman and Donovan, 1995). Our results demonstrate that the cataleptic behavior induced by dopamine D2 antagonists is strongly reduced in NGFI-B-deficient mice. The residual catalepsy observed in the NGFI-B (−/−) mice after high doses of haloperidol may result from the interaction of this drug with other amine receptor subtypes in the brain (Leysen et al, 1993). Interestingly, the level of dopamine D2 receptors is significantly increased in the StDL in NGFI-B KO mice suggesting that NGFI-B might also act upstream of D2 receptors. The effects of genetic deletion of NGFI-B are specific to dopamine D2-mediated pathways since the catalepsy induced by SCH 23390, a dopamine D1 antagonist, and D1 receptor density are not affected.
At biochemical levels, several studies have shown that administration of neuroleptics modulates a number of gene transcripts in the CNS. The two main families of transcripts studied so far include Fos and striatal neuropeptides such as enkephalin and neurotensin (Herdegen and Leah, 1998; Steiner and Gerfen, 1998). The effect of neuroleptics on opioid neuropeptide expression is seen as an adaptative phenomenon to re-establish the normal activity of dopamine systems (Steiner and Gerfen, 1998), whereas it has been suggested that neurotensin is required for the activation of specific populations of striatal neurons by typical antipsychotics (Dobner et al, 2001). Our results demonstrate that the genetic deletion of NGFI-B also interferes with the effect of haloperidol on striatal enkephalin and neurotensin expression. These two transcripts have been associated with the dopamine D2 receptor expressing cells in the striatum (Gerfen et al, 1990; Le Moine and Bloch, 1995; Augood et al, 1997). This is in agreement with the fact that catalepsy induced by D2 receptor antagonists but not D1 antagonists, is strongly reduced in NGFI-B-deficient mice. These results are also consistent with our previous data showing that haloperidol-induced NGFI-B expression is restricted to the subpopulation of striatal cells expressing the enkephalin and neurotensin transcripts (Beaudry et al, 2000). A specific association of NGFI-B with dopamine D2-mediated pathways is also supported by the fact that haloperidol-induced upregulation of c-fos mRNA is not affected by the genetic deletion of NGFI-B.
At the present time, it is difficult to pinpoint cellular events that might explain the absence of cataleptic behavior and striatal neuropeptide upregulation in NGFI-B-deficient mice. We have previously shown that acute haloperidol strongly increased NGFI-B mRNA levels in the rat forebrain (Beaudry et al, 2000), but the intracellular signaling events triggering by neuroleptics and leading to modulation of NGFI-B are unknown. It has been shown in vitro that NGFI-B is a direct target of kinases associated with G protein-coupled receptors intracellular signaling such as cyclic AMP (cAMP) and MAPK pathways (Kovalovsky et al, 2002; Slagsvold et al, 2002; Maira et al, 2003). Since activation of the dopamine D2 receptor normally reduces cAMP levels and striatal levels of NGFI-B (Gervais et al, 1999), blockade of the D2 receptor by neuroleptics may increase or release inhibition of cAMP-dependent protein kinase activity (PKA) and result in an upregulation NGFI-B expression (Beaudry et al, 2000). Also, we cannot exclude that post-translational modification (phosphorylation), which greatly affect NGFI-B activity (Maira et al, 2003), of pre-existing NGFI-B or de novo haloperidol-induced NGFI-B may play a role in the effects observed here. Other signaling pathways such as those implicating phospholipase C, intracellular calcium stores and protein kinase C (PKC) might also be involved (see Hernandez-Lopez et al, 2000). In addition, modulation of NGFI-B expression might also be indirect through modulation of glutamate neurotransmission by neuroleptic drugs (Leveque et al, 2000). Additional investigations are needed in order to identify the cellular events involved in the modulation of NGFI-B levels by neuroleptics.
The present results also indicate that retinoids might be involved in behavioral and biochemical effects of neuroleptics. The RXRγ1 isoform is by far the most abundant retinoid receptor expressed in the adult StDL (Zetterström et al, 1999; Langlois et al, 2001) and therefore, the effects of retinoid ligands are likely mediated through interaction with this receptor isoform. We show here that haloperidol administration strongly increase the percentage of colocalization of NGFI-B and RXRγ1 transcripts in the striatum. In addition, coadministration with haloperidol of two retinoid ligands, 9-cis RA and DHA (Heyman et al, 1992; de Urquiza et al, 2000), dose-dependently reduced or suppressed haloperidol-induced catalepsy and enkephalin mRNA upregulation. Although the enkephalin gene promoter contains a retinoic acid responsive element (RARE) (Chan et al, 1997), the activity of NGFI-B on the enkephalin promoter activity is not known. Interestingly, it has been previously shown that genetic ablation (KO) of the RXRγ gene also produces a blunted cataleptic behavior in response to dopamine antagonist (Saga et al, 1999) and an altered enkephalin gene expression in the striatum (Krezel et al, 1998). Based on these considerations, it is tempting to suggest that both NGFI-B and RXR, possibly as a transcriptional complex, may be involved in the signaling cascade induced by haloperidol. Indeed, it has been shown that NGFI-B and RXR can form an heterodimer that is active on transcription (Vivat et al, 1997; Castillo et al, 1998) and that 9-cis RA strongly enhances the transcriptional activity of the NGFI-B/RXR heterodimer in vitro (Heyman et al, 1992; Vivat et al, 1997).
The acute effect of retinoid ligands on haloperidol-induced catalepsy may be too rapid to support an alteration of retinoid receptor-dependent transcriptional activities. However, the effects of retinoid ligands on haloperidol-induced catalepsy is somewhat delayed and started to become apparent only 90 min after the injection of the drugs, whereas catalepsies induced by haloperidol alone or in combination with retinoid ligands are indistinguishable at earlier time points (30 and 60 min, see Figure 6a, c). On the other hand, recent evidence suggests that some steroids normally interacting with nuclear receptors also have rapid effects via a direct interaction with protein kinase activities in the cell cytoplasm (Nilsson et al, 2001). For example, a recently developed synthetic retinoid, 6-[3-(1-adamantyl)-4-hydroxyphenyl]-2-naphthalenecarboxylic acid (AHPN), can exert its cell cycle arrest and apoptotic activity by a signaling pathway independent of retinoid receptor activation (Dawson et al, 2001). Thus, further experiments will be necessary in order to identify the exact mechanism involved in the effects of 9-cis RA and DHA in the present paradigm.
Many components involved in the metabolic pathway of all-trans and 9-cis RA are missing in the adult striatum. For example, the RA-synthesizing retinaldehyde dehydrogenase enzymes are poorly expressed in the fully developed striatum (Wagner et al, 2002). The cellular retinal-binding proteins (CRBP) are not expressed in the striatum of adult rodents and the cellular retinoic acid-binding protein (CRABP) is only expressed in cholinergic neurons of the striatum and not in medium spiny neurons (Zetterström et al, 1999). These observations are not consistent with an active ligand-dependent role of retinoic acid derivatives in those cells. Thus, a ligand-independent transcriptional activity of the NGFI-B/RXR complex that is readily formed after haloperidol-induced NGFI-B mRNA levels in the striatum (Beaudry et al, 2000) may be involved. Indeed, NGFI-B possesses an uncommonly potent activation function-1 (AF-1) domain that is essential for ligand-independent activation of gene expression, cofactor recruitment and interaction with RXR isoforms (Wansa et al, 2002). Thus, we can hypothesize that addition of a RXR agonist (9-cis RA or DHA) modifies the transcriptional activity of the complex (for a ligand-dependent activity) that interferes with haloperidol-induced effects. Such a twist from constitutive to ligand-induced activity can be observed in vitro after specific mutations in the ligand-binding pocket of RXR (Vivat et al, 1997).
In lymphocyte, NGFI-B (Nur77; NR4A1) plays an important role as a proapoptotic factor, but no effect of the KO on the activity of immune cells has been observed (Liu et al, 1994; Woronicz et al, 1994). In fact, it appears that the absence of NGFI-B is totally compensated by Nor-1 (Lee et al, 1995; Cheng et al, 1997). The Nor-1 mRNA is also expressed in the striatum and, as NGFI-B its expression can be increased by acute haloperidol administration (Figure 3) (Werme et al, 2000). In the NGFI-B-deficient mice, the effect of haloperidol on Nor-1 mRNA levels is significantly higher compared to WT mice. This could indicate that some compensatory phenomenon may have developed in the NGFI-B-deficient mice. However, the effect of this putative redundancy by Nor-1 over NGFI-B activity has no apparent effect on the biochemical and behavioral components analyzed here. In addition, haloperidol-induced cataleptic response is preserved in Nor-1 KO mice (unpublished data, in collaboration with Dr Yves Labelle, Saint-Francois d'Assise Research Center, Quebec, Canada). Thus, it appears that unlike in the immune system, NGFI-B and Nor-1 expressed in the striatum have distinct functions. It has been shown that Nor-1, unlike NGFI-B (Nur77) and Nurr1, cannot form heterodimers with members of the RXR family (Paulsen et al, 1995; Zetterström et al, 1996). This reinforces the possibility of an involvement of an NGFI-B/RXR complex in the effects investiged here.
Collectively the present set of data indicate, for the first time, an involvement of NGFI-B and retinoids in a signaling cascade triggered upon administration of dopamine D2 receptor antagonists. More specifically, they suggest that NGFI-B and retinoids are involved in the generation of acute EPS (parkinsonism) and enkephalin opioid gene expression induced by a conventional neuroleptic. In addition, our data suggest that retinoid ligands might be used to prevent acute EPS induced by a typical neuroleptic. Other functions associated with the nucleus accumbens and the StDM (more limbic areas) may also be associated with an NGFI-B activity, since similar alterations of haloperidol-induced biochemical effects are observed in those structures in the NGFI-B-deficient mice (Table 1). The role of retinoids in the development of the CNS is well known (Maden, 2002) and molecular and genetic approaches have previously suggested an association of retinoid genetic markers and vulnerability to schizophrenia (Goodman, 1998; LaMantia, 1999). The present data indicate, for the first time, that retinoids (a vitamin A derivative and an ω-3 PUFA) and nuclear receptors may be involved in neuroleptic-mediated actions in fully developed animals. More experiments are needed in order to fully understand the interaction between cell surface dopamine receptors and ligand-activated transcription factors (nuclear receptors). Nevertheless, the present results suggest that other therapeutic targets (NGFI-B and retinoid receptors) may exist to improve conventional neuroleptic efficacy and reduce EPS.
References
Allison DB, Casey DE (2001). Antipsychotic-induced weight gain: a review of the literature. J Clin Psychiatry 62(Suppl 7): 22–31.
Augood SJ, Westmore K, Emson PC (1997). Phenotypic characterization of neurotensin messenger RNA-expressing cells in the neuroleptic-treated rat striatum: a detailed cellular co-expression study. Neuroscience 76: 763–774.
Beaudry G, Langlois M-C, Weppe I, Rouillard C, Lévesque D (2000). Contrasting patterns and cellular specificity of transcriptional regulation of the nuclear receptor nerve growth factor-inducible B by haloperidol and clozapine in the rat forebrain. J Neurochem 75: 1694–1702.
Castillo SO, Xiao Q, Kostrouch Z, Dozin B, Nikodem VM (1998). A divergent role of COOH-terminal domains in Nurr1 and Nur77 transactivation. Gene Exp 7: 1–12.
Chambon P (1996). A decade of molecular biology of retinoic acid receptors. FASEB J 10: 940–954.
Chan RM, Stewart MJ, Crabb DW (1997). A direct repeat (DR-1) element in the first exon modulates transcription of the preproenkephalin A gene. Brain Res Mol Brain Res 45: 50–58.
Cheng LE, Chan FK, Cado D, Winoto A (1997). Functional redundancy of the Nur77 and Nor-1 orphan steroid receptors in T-cell apoptosis. EMBO J 16: 1865–1875.
Crawford PA, Sadovsky Y, Woodson K, Lee SL, Milbrandt J (1995). Adrenocortical function and regulation of the steroid 21-hydroxylase gene in NGFI-B-deficient mice. Mol Cell Biol 15: 4331–4336.
Dawson MI, Hobbs PD, Peterson VJ, Leid M, Lange CW, Feng K-C et al (2001). Apoptosis induction in cancer cells by a novel analogue of 6-[3-(1-adamantyl)-4-hydroxyphenyl]-2-naphthalene-carboxylic acid lacking retinoid receptor transcriptional activation activity. Cancer Res 61: 4723–4730.
de Urquiza AM, Liu S, Sjöberg M, Zetterström RH, Griffiths W, Sjövall J et al (2000). Docosahexaenoic acid, a ligand for the retinoid X receptor in mouse brain. Science 290: 2140–2144.
Dobner PR, Fadel J, Deitemeyer N, Carraway RE, Deutch AY (2001). Neurotensin-deficient mice show altered responses to antipsychotic drugs. Proc Natl Acad Sci USA 98: 8048–8053.
Egea PF, Mitschler A, Moras D (2002). Molecular recognition of agonist ligands by RXRs. Mol Endocrinol 16: 987–997.
Gerfen CR, Mahan LC, Susel Z, Chase TN, Monsma Jr FJ, Sibley DR (1990). D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science 250: 1429–1432.
Gervais J, Soghomonian J-J, Richard D, Rouillard C (1999). Dopamine and serotonin interactions in the modulation of the expression of the immediate-early transcription factor, nerve growth factor-inducible B, in the striatum. Neuroscience 91: 1045–1054.
Goodman AB (1998). Three independent lines of evidence suggest retinoids as causal to schizophrenia. Proc Natl Acad Sci USA 95: 7240–7244.
Hazel TG, Nathans D, Lau LF (1988). A gene inducible by serum growth factors encodes a member of the steroid and thyroid hormone receptor superfamily. Proc Natl Acad Sci USA 85: 8444–8448.
Herdegen T, Leah JD (1998). Inducible and constitutive transcription factors in the mammalian nervous system: control of gene expression by Jun, Fos and Krox, and CREB/ATF proteins. Brain Res Brain Res Rev 28: 370–490.
Hernandez-Lopez S, Tkatch T, Perez-Garci E, Galarraga E, Bargas J, Hamm H et al (2000). D2 dopamine receptors in striatal medium spiny neurons reduce L-type Ca2+ currents and excitability via a novel PLCβ1-IP3-calcineurin-signaling cascade. J Neurosci 20: 8987–8995.
Heyman RA, Mangelsdorf DJ, Dyck JA, Stein RB, Eichele G, Evans RM et al (1992). 9-cis retinoic acid is a high affinity ligand for the retinoid X receptor. Cell 68: 397–406.
Hiroi N, Graybiel AM (1996). Atypical and typical neuroleptic treatments induce distinct programs of transcription factor expression in the striatum. J Comp Neurol 374: 70–83.
Hoffman DC, Donovan H (1995). Catalepsy as a rodent model for detecting antipsychotic drugs with extrapyramidal side effect liability. Psychopharmacology (Berl) 120: 128–133.
Kane JM (2001). Extrapyramidal side effects are unacceptable. Eur Neuropsychopharmacol 11: S397–S403.
Kovalovsky D, Refojo D, Liberman AC, Hochbaum D, Paez-Pereda M, Coso OA et al (2002). Activation and induction of NUR77/NURR1 in corticotrophs by CRH/cAMP: involvement of calcium, protein kinase A, and MAPK pathways. Mol Endocrinol 16: 1638–1651.
Krezel W, Ghyselinck N, Samad TA, Dupé V, Kastner P, Borrelli E et al (1998). Impaired locomotion and dopamine signaling in retinoid receptor mutant mice. Science 279: 863–867.
Krezel W, Kastner P, Chambon P (1999). Differential expression of retinoid receptors in the adult mouse central nervous system. Neuroscience 89: 1291–1300.
LaMantia A-S (1999). Forebrain induction, retinoic acid, and vulnerability to schizophrenia: insights from molecular and genetic analysis in developing mice. Biol Psychiatry 46: 19–30.
Langlois M-C, Beaudry G, Zekki H, Rouillard C, Lévesque D (2001). Impact of antipsychotic drug administration on the expression of nuclear receptors in the neocortex and striatum of the rat brain. Neuroscience 106: 117–128.
Le Moine C, Bloch B (1995). D1 and D2 dopamine receptor gene expression in the rat striatum: sensitive cRNA probes demonstrate prominent segregation of D1 and D2 mRNAs in distinct neuronal populations of the dorsal and ventral striatum. J Comp Neurol 355: 418–426.
Lee SL, Wesselschmidt RL, Linette GP, Kanagawa O, Russell JH, Milbrandt J (1995). Unimpaired thymic and peripheral T cell death in mice lacking the nuclear receptor NGFI-B (Nur77). Science 269: 532–535.
Leveque J-C, Macias W, Rajadhyaksha A, Carlson RR, Barczak A, Kang S et al (2000). Intracellular modulation of NMDA receptor function by antipsychotic drugs. J Neurosci 20: 4011–4020.
Leysen JE, Janssen PM, Schotte A, Luyten WH, Megens AA (1993). Interaction of antipsychotic drugs with neurotransmitter receptor sites in vitro and in vivo in relation to pharmacological and clinical effects: role of 5-HT2 receptors. Psychopharmacology (Berl) 112: S40–S54.
Liu Z-G, Smith SW, McLaughlin KA, Schwartz LM, Osborne BA (1994). Apoptotic signals delivered through the T-cell receptor of a T-cell hybrid require the immediate-early gene Nur77. Nature 367: 281–284.
Maden M (2002). Retinoid signalling in the development of the central nervous system. Nat Rev Neurosci 3: 843–853.
Maira M, Martens C, Batsché E, Gauthier Y, Drouin J (2003). Dimer-specific potentiation of NGFI-B (Nur77) transcriptional activity by the protein kinase A pathway and AF-1-dependent coactivator recruitment. Mol Cell Biol 23: 763–776.
Maltais A, Labelle Y (2000). Structure and expression of the mouse gene encoding the orphan nuclear receptor TEC. DNA Cell Biol 19: 121–130.
Meltzer HY (1995). Atypical antipsychotic drugs. In: Bloom FE, Kupfer DJ (eds). Psychopharmacology: The Fourth Generation of Progress. Raven Press: New York. pp 1277–1286.
Milbrandt J (1988). Nerve growth factor induces a gene homologous to the glucocorticoid receptor gene. Neuron 1: 183–188.
Nilsson S, Makela S, Treuter E, Tujague M, Thomsen J, Andersson G et al (2001). Mechanisms of estrogen action. Physiol Rev 81: 1535–1565.
Paulsen RF, Granas K, Johnsen H, Rolseth V, Sterri S (1995). Three related brain nuclear receptors, NGFI-B, Nurr1, and NOR-1, as transcriptional activators. J Mol Neurosci 6: 249–255.
Perlmann T, Jansson L (1995). A novel pathway for vitamin A signaling mediated by RXR heterodimerization with NGFI-B and NURR1. Genes Dev 9: 769–782.
Saga Y, Kobayashi M, Ohta H, Murai N, Nakai N, Oshima M et al (1999). Impaired extrapyramidal function caused by the targeted disruption of retinoid X receptor RXRγ1 isoform. Genes Cells 4: 219–228.
Seeman P (1995). Dopamine receptors: clinical correlates. In: Bloom FE, Kupfer DJ (eds). Psychopharmacology: The Fourth Generation of Progress. Raven Press: New York. pp 295–302.
Slagsvold HH, Ostvold A-C, Fallgren AB, Paulsen RE (2002). Nuclear receptor and apoptosis initiator NGFI-B is a substrate for kinase ERK2. Biochem Biophys Res Commun 291: 1146–1150.
Steiner H, Gerfen CR (1998). Role of dynorphin and enkephalin in the regulation of striatal output pathways and behavior. Exp Brain Res 123: 60–76.
Tremblay M, Rouillard C, Lévesque D (1999). Dopamine D3 receptor antisense administration reduces basal c-fos and NGFI-B mRNA levels in the rat forebrain. Synapse 32: 51–57.
Vivat V, Zechel C, Wurtz J-M, Bourguet W, Kagechika H, Umemiya H et al (1997). A mutation mimicking ligand-induced conformational change yields a constitutive RXR that senses allosteric effects in heterodimers. EMBO J 16: 5697–5709.
Wagner E, Luo T, Dräger UC (2002). Retinoic acid synthesis in the postnatal mouse brain marks distinct developmental stages and functional systems. Cereb Cortex 12: 1244–1253.
Wansa KD, Harris JM, Muscat GE (2002). The activation function-1 domain of Nur77/NR4A1 mediates trans-activation, cell specificity, and coactivator recruitment. J Biol Chem 277: 33001–33011.
Werme M, Ringholm A, Olson L, Brené S (2000). Differential patterns of induction of NGFI-B, Nor-1 and c-fos mRNAs in striatal subregions by haloperidol and clozapine. Brain Res 863: 112–119.
Woronicz JD, Calnan B, Ngo V, Winoto A (1994). Requirement for the orphan steroid receptor Nur77 in apoptosis of T-cell hybridomas. Nature 367: 277–281.
Zetterström RH, Lindqvist E, de Urquiza AM, Tomac A, Eriksson U, Perlmann T et al (1999). Role of retinoids in the CNS: differential expression of retinoid binding proteins and receptors and evidence for presence of retinoic acid. Eur J Neurosci 11: 407–416.
Zetterström RH, Solomin L, Mitsiadis T, Olson L, Perlmann T (1996). Retinoid X receptor heterodimerization and developmental expression distinguish the orphan nuclear receptors NGFI-B, Nurr1, and Nor1. Mol Endocrinol 10: 1656–1666.
Acknowledgements
We thank Dr Pierre-Paul Rompré and Pierre Blanchet for very stimulating and helpful discussions and Dr Yves Labelle from the SFA Research Center (Québec, Canada) for generously providing us with Nor-1 knockout mice. DL holds a scholarship from the Fonds de la Recherche en Santé du Québec (FRSQ) and IE holds a fellowship from the Laval University Neuroscience Research Center.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Éthier, I., Beaudry, G., St-Hilaire, M. et al. The Transcription Factor NGFI-B (Nur77) and Retinoids Play a Critical Role in Acute Neuroleptic-Induced Extrapyramidal Effect and Striatal Neuropeptide Gene Expression. Neuropsychopharmacol 29, 335–346 (2004). https://doi.org/10.1038/sj.npp.1300318
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.npp.1300318
Keywords
This article is cited by
-
Paliperidone and aripiprazole differentially affect the strength of calcium-secretion coupling in female pituitary lactotrophs
Scientific Reports (2015)
-
Patrolling monocytes play a critical role in CX3CR1-mediated neuroprotection during excitotoxicity
Brain Structure and Function (2015)
-
The transcription factors Nur77 and retinoid X receptors participate in amphetamine-induced locomotor activities
Psychopharmacology (2009)
