
Featured
-
-
Article
| Open Access Allopolyploid origin and diversification of the Hawaiian endemic mints
Allopolyploid origin and diversification of the Hawaiian endemic mints
Hawaiian endemic mints represent the second largest plant radiation in the archipelago. Here, the authors present a reference genome and numerous resequenced individuals to uncover evidence for polyploidy, geographic speciation and localized hybridization underlying diversification in this lineage
- Crystal M. Tomlin
- , Sitaram Rajaraman
- & Charlotte Lindqvist
-
Article
| Open Access The recent rapid expansion of multidrug resistant Ural lineage Mycobacterium tuberculosis in Moldova
The recent rapid expansion of multidrug resistant Ural lineage Mycobacterium tuberculosis in Moldova
Chitwood et al. report on the rapid expansion of a Ural-lineage multidrug resistant strain of Mycobacterium tuberculosis in Moldova. This strain has an estimated reproduction number more than two times greater than otherwise similar drug susceptible strains.
- Melanie H. Chitwood
- , Caroline Colijn
- & Benjamin Sobkowiak
-
Article
| Open Access Genomic epidemiology reveals geographical clustering of multidrug-resistant Escherichia coli ST131 associated with bacteraemia in Wales
Genomic epidemiology reveals geographical clustering of multidrug-resistant Escherichia coli ST131 associated with bacteraemia in Wales
Escherichia coli ST131 is a globally dominant multidrug resistant clone associated with high rates of recurring urinary tract infections. In this genomic epidemiology study, the authors describe the evolution, population structure, and antimicrobial resistance in 142 E. coli ST131 samples from Wales, UK.
- Rhys T. White
- , Matthew J. Bull
- & Scott A. Beatson
-
Article
| Open Access Systematic review and integrated data analysis reveal diverse pangolin-associated microbes with infection potential
Systematic review and integrated data analysis reveal diverse pangolin-associated microbes with infection potential
The diversity and spillover potential of pangolin-associated microbes are not fully understood. Here, the authors describe the distribution and spectrum of reported pangolin microbes by integrating data from multiple sources and assess their potential to emerge as human pathogens.
- Run-Ze Ye
- , Xiao-Yang Wang
- & Wu-Chun Cao
-
Article
| Open Access A genomic appraisal of invasive Salmonella Typhimurium and associated antibiotic resistance in sub-Saharan Africa
A genomic appraisal of invasive Salmonella Typhimurium and associated antibiotic resistance in sub-Saharan Africa
Invasive Salmonella Typhimurium bloodstream infection causes a significant public health burden in sub-Saharan Africa. Here, the authors analyse whole genome sequences of 1,302 S. Typhimurium isolates from Africa and describe its evolution, geographic spread, and antimicrobial resistance characteristics.
- Sandra Van Puyvelde
- , Tessa de Block
- & Octavie Lunguya
-
Article
| Open Access COMPASS: joint copy number and mutation phylogeny reconstruction from amplicon single-cell sequencing data
COMPASS: joint copy number and mutation phylogeny reconstruction from amplicon single-cell sequencing data
Understanding the evolution of a tumor is important for predicting its resistance to treatment. This paper presents a new computational method, COMPASS, for inferring the joint phylogeny of single nucleotide variants and copy number alterations from targeted scDNAseq data.
- Etienne Sollier
- , Jack Kuipers
- & Katharina Jahn
-
Article
| Open Access Increased interregional virus exchange and nucleotide diversity outline the expansion of chikungunya virus in Brazil
Increased interregional virus exchange and nucleotide diversity outline the expansion of chikungunya virus in Brazil
Chikungunya virus is endemic in Brazil and cases have been rapidly increasing in recent years. Here, the authors describe the expansion of a genomic surveillance program across the country allowing them to characterise the emergence and dispersal of two distinct subclades mainly seeded from the north eastern region.
- Joilson Xavier
- , Luiz Carlos Junior Alcantara
- & Marta Giovanetti
-
Article
| Open Access Integrating full and partial genome sequences to decipher the global spread of canine rabies virus
Integrating full and partial genome sequences to decipher the global spread of canine rabies virus
Although pathogen whole genome sequencing is becoming more common, for many pathogens far more partial sequences are available. In this study, the authors develop a phylogenetic pipeline to efficiently combine whole and partial viral genome sequences and demonstrate its application using rabies virus sequences.
- Andrew Holtz
- , Guy Baele
- & Anna Zhukova
-
Article
| Open Access Parallel and convergent genomic changes underlie independent subterranean colonization across beetles
Parallel and convergent genomic changes underlie independent subterranean colonization across beetles
The genomic underpinnings of cave-related phenotypes are underexplored. Here, the authors investigate adaptation to underground life in cave beetle lineages using transcriptomic and genomic data, finding both parallel and convergent changes in six independent episodes of subterranean colonization.
- Pau Balart-García
- , Leandro Aristide
- & Rosa Fernández
-
Article
| Open Access A global genomic analysis of Salmonella Concord reveals lineages with high antimicrobial resistance in Ethiopia
A global genomic analysis of Salmonella Concord reveals lineages with high antimicrobial resistance in Ethiopia
Authors carry out a longitudinal genomic analysis of Salmonella enterica serovar Concord isolates from various geographical locations, to reconstruct population diversity, evolution and antimicrobial resistance distribution.
- Wim L. Cuypers
- , Pieter Meysman
- & Sandra Van Puyvelde
-
Article
| Open Access Identification of a covert evolutionary pathway between two protein folds
Identification of a covert evolutionary pathway between two protein folds
Protein secondary structures–α-helices and β-sheets–are generally assumed to be fixed over evolutionary history. By leveraging sequence information and sensitive statistical techniques, this work proposes that secondary structures in naturally occurring DNA-binding proteins switched in response to stepwise mutation.
- Devlina Chakravarty
- , Shwetha Sreenivasan
- & Lauren L. Porter
-
Article
| Open Access Decoupling body shape and mass distribution in birds and their dinosaurian ancestors
Decoupling body shape and mass distribution in birds and their dinosaurian ancestors
Here, the authors track the evolution of mass distribution through bird evolution challenging suggested coupling between body shape and centre-of-mass position, and instead showing that crouched bipedalism evolved after powered flight.
- Sophie Macaulay
- , Tatjana Hoehfurtner
- & Karl T. Bates
-
Article
| Open Access Reconstructing clonal tree for phylo-phenotypic characterization of cancer using single-cell transcriptomics
Reconstructing clonal tree for phylo-phenotypic characterization of cancer using single-cell transcriptomics
The functional changes of individual clones in single cell RNA sequencing (scRNA-seq) data remain elusive. Here, the authors develop PhylEx that integrates bulk genomics data with co-occurrences of mutations revealed by scRNA-seq data and apply it to high-grade serous ovarian cancer cell line and breast cancer datasets.
- Seong-Hwan Jun
- , Hosein Toosi
- & Jens Lagergren
-
Matters Arising
| Open Access Reply to: Available data do not rule out Ctenophora as the sister group to all other Metazoa
Reply to: Available data do not rule out Ctenophora as the sister group to all other Metazoa
- Anthony K. Redmond
- & Aoife McLysaght
-
Matters Arising
| Open Access Available data do not rule out Ctenophora as the sister group to all other Metazoa
Available data do not rule out Ctenophora as the sister group to all other Metazoa
- Nathan V. Whelan
- & Kenneth M. Halanych
-
Article
| Open Access Dynamics of extended-spectrum cephalosporin resistance genes in Escherichia coli from Europe and North America
Dynamics of extended-spectrum cephalosporin resistance genes in Escherichia coli from Europe and North America
Extended-spectrum cephalosporin resistance genes in Escherichia coli have spread worldwide. Here, the authors dissect the emergence and distribution of these genes over time, and across geographic location and host species, to better understand their dynamics and mechanisms of transmission.
- Roxana Zamudio
- , Patrick Boerlin
- & Alison E. Mather
-
Article
| Open Access Ordovician opabiniid-like animals and the role of the proboscis in euarthropod head evolution
Ordovician opabiniid-like animals and the role of the proboscis in euarthropod head evolution
Here, the authors describe two opabiniid-like euarthropods with anterior proboscises from the Middle Ordovician Castle Bank Biota, Wales, UK. Phylogenetic analysis suggests that these specimens may be sister to radiodonts and deuteropods.
- Stephen Pates
- , Joseph P. Botting
- & Joanna M. Wolfe
-
Article
| Open Access Common evolutionary origin of acoustic communication in choanate vertebrates
Common evolutionary origin of acoustic communication in choanate vertebrates
Here, the authors record acoustic communication in 53 species commonly considered non-vocal and reconstruct acoustic communication as originating 407 million years ago.
- Gabriel Jorgewich-Cohen
- , Simon William Townsend
- & Marcelo R. Sánchez-Villagra
-
Article
| Open Access Protein language models trained on multiple sequence alignments learn phylogenetic relationships
Protein language models trained on multiple sequence alignments learn phylogenetic relationships
Protein language models taking multiple sequence alignments as inputs capture protein structure and mutational effects. Here, the authors show that these models also encode phylogenetic relationships, and can disentangle correlations due to structural constraints from those due to phylogeny.
- Umberto Lupo
- , Damiano Sgarbossa
- & Anne-Florence Bitbol
-
Article
| Open Access Past and present giant viruses diversity explored through permafrost metagenomics
Past and present giant viruses diversity explored through permafrost metagenomics
Although giant viruses are abundant in aquatic environments, less is known about giant viruses in soil. Here, the authors use permafrost metagenomics to reveal giant virus diversity and heterogeneity, as well as gene transfers between viruses from different families.
- Sofia Rigou
- , Sébastien Santini
- & Matthieu Legendre
-
Article
| Open Access Phylogeographic analysis reveals an ancient East African origin of human herpes simplex virus 2 dispersal out-of-Africa
Phylogeographic analysis reveals an ancient East African origin of human herpes simplex virus 2 dispersal out-of-Africa
There are competing hypotheses for human herpes simplex virus 2’s migration out-of-Africa. Here, the authors sequence 65 new herpes simplex virus 2 genomes with a focus on under-sampled sub-Saharan African countries, suggesting an Eastern African origin for global dispersal the virus between 22-29 thousand years ago.
- Jennifer L. Havens
- , Sébastien Calvignac-Spencer
- & Joel O. Wertheim
-
Article
| Open Access Transcontinental spread and evolution of Mycobacterium tuberculosis W148 European/Russian clade toward extensively drug resistant tuberculosis
Transcontinental spread and evolution of Mycobacterium tuberculosis W148 European/Russian clade toward extensively drug resistant tuberculosis
An outbreak of the multidrug-resistant Mycobacterium tuberculosis lineage W148 has spread widely across Russia, Central Asia and Europe. Here, the authors use whole genome sequences of ~700 isolates of this lineage collected over ~20 years to analyze its spread, evolution of drug resistance, and impact of compensatory mutations.
- Matthias Merker
- , Jean-Philippe Rasigade
- & Thierry Wirth
-
Article
| Open Access Clonal diversification and histogenesis of malignant germ cell tumours
Clonal diversification and histogenesis of malignant germ cell tumours
The molecular characterisation of germ cell tumours (GCT) is necessary to understand their development and histological diversification. Here, the authors use whole-genome and transcriptome sequencing of GCTs across distinct histologies to reveal their somatic evolution and clonal diversification, as well as identify several putative biomarkers for treatment stratification.
- Thomas R. W. Oliver
- , Lia Chappell
- & Sam Behjati
-
Article
| Open Access A Bayesian approach to infer recombination patterns in coronaviruses
A Bayesian approach to infer recombination patterns in coronaviruses
Genetic recombination can confound standard phylogenetic approaches. Here, the authors present a method to reconstruct virus recombination networks, and show the importance of recombination in shaping the ongoing evolution of SARS-like, MERS and 3 human seasonal coronaviruses.
- Nicola F. Müller
- , Kathryn E. Kistler
- & Trevor Bedford
-
Article
| Open Access Deep learning from phylogenies to uncover the epidemiological dynamics of outbreaks
Deep learning from phylogenies to uncover the epidemiological dynamics of outbreaks
Widely applicable, accurate and fast inference methods in phylodynamics are needed to fully profit from the richness of genetic data in uncovering the dynamics of epidemics. Here, the authors develop a likelihood-free, simulation-based deep learning approach.
- J. Voznica
- , A. Zhukova
- & O. Gascuel
-
Article
| Open Access Contribution of low population immunity to the severe Omicron BA.2 outbreak in Hong Kong
Contribution of low population immunity to the severe Omicron BA.2 outbreak in Hong Kong
Hong Kong experienced a severe wave of SARS-CoV-2 in early 2022. Here, the authors use genomic and serosurveillance data and show that this wave was dominated by the Omicron BA.2 sublineage, and that low protective immunity, particularly in older age groups, contributed to its severity.
- Lin-Lei Chen
- , Syed Muhammad Umer Abdullah
- & Kelvin Kai-Wang To
-
Article
| Open Access Optimising genomic approaches for identifying vancomycin-resistant Enterococcus faecium transmission in healthcare settings
Optimising genomic approaches for identifying vancomycin-resistant Enterococcus faecium transmission in healthcare settings
Vancomycin-resistant Enterococcus faecium is an important healthcare-associated pathogen and genomic analyses could inform targeted interventions. Here, the authors optimise an analysis pipeline for identification of putative transmission events using core genome multilocus sequence type clustering and split kmer analysis.
- Charlie Higgs
- , Norelle L. Sherry
- & Benjamin P. Howden
-
Article
| Open Access The rise of grasslands is linked to atmospheric CO2 decline in the late Palaeogene
The rise of grasslands is linked to atmospheric CO2 decline in the late Palaeogene
A better understanding of how grasslands have responded to past environmental changes will help predict the outcomes of future changes. This study explores past climatic fluctuations and shifts in the diversification rate of grasses and daisies, finding strong evidence for a simultaneous increase in their diversification rates following a reduction of atmospheric CO2 in the Cenozoic.
- Luis Palazzesi
- , Oriane Hidalgo
- & Sebastian Höhna
-
Article
| Open Access Co-evolution based machine-learning for predicting functional interactions between human genes
Co-evolution based machine-learning for predicting functional interactions between human genes
With the rise in number of eukaryotic species being fully sequenced, large scale phylogenetic profiling can give insights on gene function, Here, the authors describe a machine-learning approach that integrates co-evolution across eukaryotic clades to predict gene function and functional interactions among human genes.
- Doron Stupp
- , Elad Sharon
- & Yuval Tabach
-
Article
| Open Access Rapid incidence estimation from SARS-CoV-2 genomes reveals decreased case detection in Europe during summer 2020
Rapid incidence estimation from SARS-CoV-2 genomes reveals decreased case detection in Europe during summer 2020
The true number of infections from SARS-Cov-2 is unknown and believed to exceed the reported numbers by several fold. National testing policies, in particular, can strongly affect the proportion of undetected cases. Here, the authors propose a method that reconstructs incidence profiles within minutes, solely from publicly available, time-stamped viral genomes.
- Maureen Rebecca Smith
- , Maria Trofimova
- & Max von Kleist
-
Article
| Open Access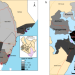 Tracking the introduction and spread of SARS-CoV-2 in coastal Kenya
Tracking the introduction and spread of SARS-CoV-2 in coastal Kenya
SARS-CoV-2 was first detected in Kenya in March 2020 and there was evidence of local transmission in the following months. Here, the authors characterise the early stages of the epidemic in coastal Kenya using phylogenetics and find evidence of multiple strain importations from international points of entry.
- George Githinji
- , Zaydah R. de Laurent
- & Charles N. Agoti
-
Article
| Open Access Genetic evidence for the association between COVID-19 epidemic severity and timing of non-pharmaceutical interventions
Genetic evidence for the association between COVID-19 epidemic severity and timing of non-pharmaceutical interventions
Estimating the effects of non-pharmaceutical interventions for COVID-19 is challenging, partly due to variations in testing. Here, the authors use viral sequence data as an alternative means of inferring intervention effects, and show that delays in implementation resulted in more severe epidemics.
- Manon Ragonnet-Cronin
- , Olivia Boyd
- & Erik Volz
-
Article
| Open Access Evidence for sponges as sister to all other animals from partitioned phylogenomics with mixture models and recoding
Evidence for sponges as sister to all other animals from partitioned phylogenomics with mixture models and recoding
Branching artefacts can confound the reconstruction of deep evolutionary relationships. Here, the authors show improvement in partitioned phylogenomic analyses when using site-heterogeneous models and amino acid recoding, including in resolving the debated phylogenetic placement of comb jellies.
- Anthony K. Redmond
- & Aoife McLysaght
-
Article
| Open Access Evolution of combinatorial diversity in trans-acyltransferase polyketide synthase assembly lines across bacteria
Evolution of combinatorial diversity in trans-acyltransferase polyketide synthase assembly lines across bacteria
Trans-acyltransferase polyketide synthases are multimodular enzymes that synthesise diverse polyketides. Here, the authors present an algorithm for the global study of their diversity, showing exchange of conserved consecutive modules as a driver of diversification, and guiding the discovery of polyketides.
- Eric J. N. Helfrich
- , Reiko Ueoka
- & Marnix H. Medema
-
Article
| Open Access A collection of bacterial isolates from the pig intestine reveals functional and taxonomic diversity
A collection of bacterial isolates from the pig intestine reveals functional and taxonomic diversity
The authors present a public collection of 117 bacterial isolates from the pig gut, including the description of 38 novel taxa. Interesting functions discovered in these organisms include a new fucosyltransferease and sactipeptide-like molecules encoded by biosynthetic gene clusters.
- David Wylensek
- , Thomas C. A. Hitch
- & Thomas Clavel
-
Article
| Open Access An investigation of irreproducibility in maximum likelihood phylogenetic inference
An investigation of irreproducibility in maximum likelihood phylogenetic inference
Replicate runs of maximum likelihood phylogenetic analyses can generate different tree topologies due to differences in parameters, such as random seeds. Here, Shen et al. demonstrate that replicate runs can generate substantially different tree topologies even with identical data and parameters.
- Xing-Xing Shen
- , Yuanning Li
- & Antonis Rokas
-
Article
| Open Access Pathways for horizontal gene transfer in bacteria revealed by a global map of their plasmids
Pathways for horizontal gene transfer in bacteria revealed by a global map of their plasmids
Plasmids can mediate gene transfer across bacterial populations. Here, the authors describe a global map of the prokaryotic plasmidome, where plasmids organize into discrete ‘plasmid taxonomic units’ based on their genomic composition and pairwise sequence identity.
- Santiago Redondo-Salvo
- , Raúl Fernández-López
- & Fernando de la Cruz
-
Article
| Open Access Single-cell lineage tracing by integrating CRISPR-Cas9 mutations with transcriptomic data
Single-cell lineage tracing by integrating CRISPR-Cas9 mutations with transcriptomic data
Lineage tracing studies combining CRISPR-Cas9 editing and scRNA-seq face several challenges and cannot integrate lineages from multiple individuals. Here the authors show that integration of mutation and expression leads to accurate lineage tree inference and enables the learning of a species-invariant lineage tree.
- Hamim Zafar
- , Chieh Lin
- & Ziv Bar-Joseph
-
Article
| Open Access Precise phylogenetic analysis of microbial isolates and genomes from metagenomes using PhyloPhlAn 3.0
Precise phylogenetic analysis of microbial isolates and genomes from metagenomes using PhyloPhlAn 3.0
The increasing amount of sequenced microbial genomes and metagenomes requires platforms for efficient integrated analysis. Here, Asnicar et al. present PhyloPhlAn 3.0, a pipeline allowing large-scale microbial genome characterization and phylogenetic contextualization at multiple levels of resolution.
- Francesco Asnicar
- , Andrew Maltez Thomas
- & Nicola Segata
-
Article
| Open Access Mapping the breast cancer metastatic cascade onto ctDNA using genetic and epigenetic clonal tracking
Mapping the breast cancer metastatic cascade onto ctDNA using genetic and epigenetic clonal tracking
Tracking tumour evolution in a patient via circulating tumour DNA (ctDNA) is complicated due to the unknown mix of fragmented alleles from different cancer lesions. Here, the authors make use of a rapid autopsy program to demonstrate how representative ctDNA profiling is of metastasis, as well as presenting methylation profiling method to track evolutionary change.
- George D. Cresswell
- , Daniel Nichol
- & Andrea Sottoriva
-
Article
| Open Access Deciphering protein evolution and fitness landscapes with latent space models
Deciphering protein evolution and fitness landscapes with latent space models
Multiple sequence alignments of proteins carry information about evolution, the protein’s fitness landscape and its stability in the face of mutations. Here, the authors demonstrate the utility of latent space models learned using variational autoencoders to infer these properties from sequences.
- Xinqiang Ding
- , Zhengting Zou
- & Charles L. Brooks III
-
Article
| Open Access Rapid evolution and biogeographic spread in a colorectal cancer
Rapid evolution and biogeographic spread in a colorectal cancer
The clonal origins of metastases and the timing of dissemination remains an open question for most cancer types. Using primary and metastatic samples taken from one colorectal cancer patient, Alves et al. use Bayesian phylogenetics to reconstruct the history of metastasis.
- Joao M. Alves
- , Sonia Prado-López
- & David Posada
-
Article
| Open Access An African Salmonella Typhimurium ST313 sublineage with extensive drug-resistance and signatures of host adaptation
An African Salmonella Typhimurium ST313 sublineage with extensive drug-resistance and signatures of host adaptation
Invasive non-typhoidal Salmonella (iNTS) infections are dominated by antibiotic resistant isolates of the sequence type (ST) 313. Here, the authors identify the ST313 sublineage II.1 in the Democratic Republic of the Congo exhibiting extensive drug resistance and genetic signatures potentially associated with host adaptation.
- Sandra Van Puyvelde
- , Derek Pickard
- & Stijn Deborggraeve
-
Article
| Open Access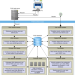 TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy
TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy
Information on type material is of fundamental importance in prokaryote taxonomy. Here, the authors develop TYGS, the Type (Strain) Genome Server, for genome-based prokaryote taxonomy and analysis using a comprehensive database of genomic and taxonomic data.
- Jan P. Meier-Kolthoff
- & Markus Göker
-
Article
| Open Access Three phylogenetic groups have driven the recent population expansion of Cryptococcus neoformans
Three phylogenetic groups have driven the recent population expansion of Cryptococcus neoformans
Cryptococcus neoformans is an opportunistic fungal pathogen which primarily affects people with immune defects including those living with HIV. Here, the authors sequence and analyze genomes of 699 isolates, and identify recent population expansion driven by three phylogenetic groups.
- P. M. Ashton
- , L. T. Thanh
- & J. N. Day
-
Article
| Open Access Model selection may not be a mandatory step for phylogeny reconstruction
Model selection may not be a mandatory step for phylogeny reconstruction
Model selection is a time-intensive step of molecular phylogenetic analysis. Here, Abadi, Azouri and colleagues show that all model selection criteria lead to similar inferences, and that for topology and ancestral sequence reconstruction, using the GTR+I+G model is as accurate.
- Shiran Abadi
- , Dana Azouri
- & Itay Mayrose
-
Article
| Open Access Single-cell mutation identification via phylogenetic inference
Single-cell mutation identification via phylogenetic inference
Cross-cell heterogeneity of genotypes can be revealed by analyzing single-cell sequencing data. Here the authors develop a tool for single-cell variant calling via phylogenetic inference, and use it to analyze cancer genomics datasets.
- Jochen Singer
- , Jack Kuipers
- & Niko Beerenwinkel
-
Article
| Open Access Broad phylogenetic analysis of cation/proton antiporters reveals transport determinants
Broad phylogenetic analysis of cation/proton antiporters reveals transport determinants
Cation/proton antiporters (CPAs) play a major role in maintaining living cells’ homeostasis and are divided in two main groups: CPA1 and CPA2. Here authors use a comprehensive evolutionary analysis of 6537 representative CPAs and reveal a sequence motif that determines central phenotypic characteristics.
- Gal Masrati
- , Manish Dwivedi
- & Nir Ben-Tal
-
Article
| Open Access Phylogenomics uncovers early hybridization and adaptive loci shaping the radiation of Lake Tanganyika cichlid fishes
Phylogenomics uncovers early hybridization and adaptive loci shaping the radiation of Lake Tanganyika cichlid fishes
Lake Tanganyika’s cichlid radiation is the main source of East African cichlid diversity. Irisarri et al. resolve its phylogenetic backbone using anchored phylogenomics and identify trans-lineage hybridization prior to major speciation bursts and adaptive loci underlying ecological innovations.
- Iker Irisarri
- , Pooja Singh
- & Axel Meyer
 Phylogenomic profiles of whole-genome duplications in Poaceae and landscape of differential duplicate retention and losses among major Poaceae lineages
Phylogenomic profiles of whole-genome duplications in Poaceae and landscape of differential duplicate retention and losses among major Poaceae lineages