
Article
|
Open Access
Featured
-
-
Comment
| Open Access Implementing community-engaged pharmacogenomics in Indigenous communities
Implementing community-engaged pharmacogenomics in Indigenous communities
Innovative pharmacogenomic approaches (genetic variation related to medication response) are needed to reduce disease and disparities in Indigenous communities. We support community-based pharmacogenomics research, inclusive of Indigenous values and priorities, to improve the health and well-being of Indigenous peoples.
- Katrina G. Claw
- , Casey R. Dorr
- & Erica L. Woodahl
-
Article
| Open Access Single-nucleus DNA sequencing reveals hidden somatic loss-of-heterozygosity in Cerebral Cavernous Malformations
Single-nucleus DNA sequencing reveals hidden somatic loss-of-heterozygosity in Cerebral Cavernous Malformations
Here the authors establish somatic loss-of-heterozygosity as a genetic underpinning of Cerebral Cavernous Malformations (CCMs): using single-nucleus DNA sequencing, they show homozygosity of chromosomes 7p and/or 7q leads to biallelic inactivation of CCM genes in resected lesions.
- Andrew K. Ressler
- , Daniel A. Snellings
- & Douglas A. Marchuk
-
Article
| Open Access A common East-Asian ALDH2 mutation causes metabolic disorders and the therapeutic effect of ALDH2 activators
A common East-Asian ALDH2 mutation causes metabolic disorders and the therapeutic effect of ALDH2 activators
A common East Asian-specific defect of an alcohol metabolizing enzyme (ALDH2) causes glucose abnormality, obesity, and fatty liver. Here, the authors show an ALDH2 activator can treat these metabolic disorders in mice.
- Yi-Cheng Chang
- , Hsiao-Lin Lee
- & Lee-Ming Chuang
-
Article
| Open Access Effect of apolipoprotein genotype and educational attainment on cognitive function in autosomal dominant Alzheimer’s disease
Effect of apolipoprotein genotype and educational attainment on cognitive function in autosomal dominant Alzheimer’s disease
PSEN1 E280A carriers develop dementia by midlife, but there is variability in disease trajectory. Cognitive decline is accelerated in E280A carriers who also have an APOE e4 allele. Educational attainment moderates the effect of APOE on cognition.
- Stephanie Langella
- , N. Gil Barksdale
- & Yakeel T. Quiroz
-
Article
| Open Access Deficit of homozygosity among 1.52 million individuals and genetic causes of recessive lethality
Deficit of homozygosity among 1.52 million individuals and genetic causes of recessive lethality
Genotypes causing pregnancy loss and perinatal mortality are depleted among living individuals and are therefore difficult to find. Here, using genetic data for 1.52 million individuals, the authors identify 25 genes with protein-altering variants exhibiting a strong deficit of homozygosity, suggesting they are essential for successful early development.
- Asmundur Oddsson
- , Patrick Sulem
- & Daniel F. Gudbjartsson
-
Article
| Open Access A computational method for cell type-specific expression quantitative trait loci mapping using bulk RNA-seq data
A computational method for cell type-specific expression quantitative trait loci mapping using bulk RNA-seq data
Detecting cell type-specific genetic effects on gene expression is challenging in bulk RNA-seq data. Here, the authors develop a method to increase power which incorporates allele-specific expression and does not transform the gene expression data.
- Paul Little
- , Si Liu
- & Wei Sun
-
Article
| Open Access Genetic architecture of spatial electrical biomarkers for cardiac arrhythmia and relationship with cardiovascular disease
Genetic architecture of spatial electrical biomarkers for cardiac arrhythmia and relationship with cardiovascular disease
The spatial and frontal QRS-T angles are electrocardiographic (ECG) predictors for arrhythmia. This work used genetic analyses to identify associated loci and pathways, and explore their relationships with other ECG traits and cardiovascular disease.
- William J. Young
- , Jeffrey Haessler
- & Patricia B. Munroe
-
Article
| Open Access Multitrait genome-wide analyses identify new susceptibility loci and candidate drugs to primary sclerosing cholangitis
Multitrait genome-wide analyses identify new susceptibility loci and candidate drugs to primary sclerosing cholangitis
The genetic basis of primary sclerosing cholangitis has only been partially uncovered. Here, the authors perform a multitrait genome-wide association study to provide insight into the genetic etiology of primary sclerosing cholangitis risk and possible therapeutic drug targets.
- Younghun Han
- , Jinyoung Byun
- & Christopher I. Amos
-
Article
| Open Access Germline TP53 mutations undergo copy number gain years prior to tumor diagnosis
Germline TP53 mutations undergo copy number gain years prior to tumor diagnosis
Li-Fraumeni syndrome (LFS) is associated with pathogenic germline TP53 variants and predisposes patients to cancer; understanding the evolution and drivers of LFS-related tumours remains crucial. Here, the authors analyse 22 LFS tumours using whole-genome sequencing and reconstruct the evolution and timing of somatic driver alterations.
- Nicholas Light
- , Mehdi Layeghifard
- & Adam Shlien
-
Article
| Open Access CRISPR-based targeted haplotype-resolved assembly of a megabase region
CRISPR-based targeted haplotype-resolved assembly of a megabase region
Low-cost targeted approach to construct haplotype-resolved assemblies is needed to facilitate population genetic studies. Here, the authors demonstrate assembling high-quality MHC haplotypes with CRISPR-based enrichment and long-read sequencings.
- Taotao Li
- , Duo Du
- & Yun Liu
-
Article
| Open Access Genome-wide association study identifies Sjögren’s risk loci with functional implications in immune and glandular cells
Genome-wide association study identifies Sjögren’s risk loci with functional implications in immune and glandular cells
The genetic architecture underlying Sjögren’s syndrome is not fully understood. Here, the authors perform a genome-wide association study to identify 10 new genetic risk regions, implicating genes involved in immune and salivary gland function.
- Bhuwan Khatri
- , Kandice L. Tessneer
- & Christopher J. Lessard
-
Article
| Open Access Rare variant analysis in eczema identifies exonic variants in DUSP1, NOTCH4 and SLC9A4
Rare variant analysis in eczema identifies exonic variants in DUSP1, NOTCH4 and SLC9A4
Genetic studies of eczema to date have mostly explored common genetic variation. Here, the authors perform a large meta-analysis for common and rare variants and discover 8 loci associated with eczema. Over 20% of the heritability of the condition is attributable to rare variants.
- Sarah Grosche
- , Ingo Marenholz
- & Young-Ae Lee
-
Article
| Open Access Incorporating functional priors improves polygenic prediction accuracy in UK Biobank and 23andMe data sets
Incorporating functional priors improves polygenic prediction accuracy in UK Biobank and 23andMe data sets
Incorporating functional information has shown promise for improving polygenic risk prediction of complex traits. Here, the authors describe polygenic prediction method LDpred-funct, and demonstrate its utility across 21 heritable traits in the UK Biobank.
- Carla Márquez-Luna
- , Steven Gazal
- & Alkes L. Price
-
Article
| Open Access Calibrated rare variant genetic risk scores for complex disease prediction using large exome sequence repositories
Calibrated rare variant genetic risk scores for complex disease prediction using large exome sequence repositories
Identifying associations of rare variants with disease is challenging due to small effect sizes, technical artefacts and population structure heterogeneity. Here, the authors present RV-EXCALIBER, a method that uses large summary-level exome data to robustly calibrate rare variant burden.
- Ricky Lali
- , Michael Chong
- & Guillaume Paré
-
Article
| Open Access KL-VS heterozygosity is associated with lower amyloid-dependent tau accumulation and memory impairment in Alzheimer’s disease
KL-VS heterozygosity is associated with lower amyloid-dependent tau accumulation and memory impairment in Alzheimer’s disease
The KL-VS haplotype of the Klotho gene has been associated with reduced risk of Alzheimer’s disease and dementia. Here the authors show an association between the KL-VS haplotype and amyloid-dependent tau accumulation using PET data.
- Julia Neitzel
- , Nicolai Franzmeier
- & Michael Ewers
-
Article
| Open Access The impact of non-additive genetic associations on age-related complex diseases
The impact of non-additive genetic associations on age-related complex diseases
Most genome-wide association studies assume an additive model, exclude the X chromosome, and use one reference panel. Here, the authors implement a strategy including non-additive models and find that the number of loci for age-related traits increases as compared to the additive model alone.
- Marta Guindo-Martínez
- , Ramon Amela
- & David Torrents
-
Article
| Open Access Genome-wide association study of smoking trajectory and meta-analysis of smoking status in 842,000 individuals
Genome-wide association study of smoking trajectory and meta-analysis of smoking status in 842,000 individuals
Genome-wide association studies (GWASs) for cigarette smoking have identified several hundred loci that account for a small proportion of the overall genetic risk. Here, the authors report a large GWAS for smoking trajectories and meta-analysis for smoking status, finding multiple plausible loci.
- Ke Xu
- , Boyang Li
- & Amy C. Justice
-
Article
| Open Access FastClone is a probabilistic tool for deconvoluting tumor heterogeneity in bulk-sequencing samples
FastClone is a probabilistic tool for deconvoluting tumor heterogeneity in bulk-sequencing samples
Multiple algorithms exist for predicting heterogeneity and clonal architecture from the bulk sequencing of tumor tissue. Here, the authors report on an algorithm, FastClone, which was developed from a DREAM challenge and show that FastClone can accurately predict clonality in simulated data and data from colon cancer.
- Yao Xiao
- , Xueqing Wang
- & Yuanfang Guan
-
Article
| Open Access Distinct genetic architectures and environmental factors associate with host response to the γ2-herpesvirus infections
Distinct genetic architectures and environmental factors associate with host response to the γ2-herpesvirus infections
Disease prognosis after infection with Kaposi’s sarcoma-associated herpesvirus and Epstein-Barr Virus is highly variable. Here the authors carry out epidemiological and genetic analysis of a Ugandan cohort and suggest complex interactions may influence pathogenesis and transmission.
- Neneh Sallah
- , Wendell Miley
- & Inês Barroso
-
Article
| Open Access Detecting sample swaps in diverse NGS data types using linkage disequilibrium
Detecting sample swaps in diverse NGS data types using linkage disequilibrium
Parallelized analysis in clinical genomics can lead to sample or data mislabelling, and could have serious downstream consequences. Here the authors present a tool to quantify sample genetic relatedness and detect such mistakes, and apply it to thousands of datasets from the ENCODE consortium.
- Nauman Javed
- , Yossi Farjoun
- & Noam Shoresh
-
Article
| Open Access Landscape of multi-nucleotide variants in 125,748 human exomes and 15,708 genomes
Landscape of multi-nucleotide variants in 125,748 human exomes and 15,708 genomes
Multi-nucleotide variants (MNV) are genetic variants in close proximity of each other on the same haplotype whose functional impact is difficult to predict if they reside in the same codon. Here, Wang et al. use the gnomAD dataset to assemble a catalogue of MNVs and estimate their global mutation rate.
- Qingbo Wang
- , Emma Pierce-Hoffman
- & Daniel G. MacArthur
-
Article
| Open Access Rare copy number variants in over 100,000 European ancestry subjects reveal multiple disease associations
Rare copy number variants in over 100,000 European ancestry subjects reveal multiple disease associations
Associations of copy number variations (CNVs) with complex traits are challenging to study because of their low frequency. Here, the authors analyse SNP array and array comparative genomic hybridization data of 100,028 individuals and report their associations with immune-related, cardiometabolic and neuropsychiatric diseases as well as cancer.
- Yun Rose Li
- , Joseph T. Glessner
- & Hakon Hakonarson
-
Article
| Open Access Functional disease architectures reveal unique biological role of transposable elements
Functional disease architectures reveal unique biological role of transposable elements
Transposable elements (TE) make up a large component of the human genome and have been shown to contribute to human diseases. Here, Hormozdiari
et al . estimate the contribution of TEs to the heritability of 41 complex traits and diseases and find enrichment of SINEs in blood traits.- Farhad Hormozdiari
- , Bryce van de Geijn
- & Alkes L. Price
-
Article
| Open Access Genome-wide association study of medication-use and associated disease in the UK Biobank
Genome-wide association study of medication-use and associated disease in the UK Biobank
An understanding of the genetic variants associated with medication use may shed light on the underlying biological pathways of disease, and aid in drug development. Here, Wu and colleagues conduct a GWAS for self-reported medication-use in the UK Biobank, finding more than 500 independent variants and many promising leads for future work.
- Yeda Wu
- , Enda M. Byrne
- & Naomi R. Wray
-
Article
| Open Access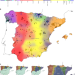 Patterns of genetic differentiation and the footprints of historical migrations in the Iberian Peninsula
Patterns of genetic differentiation and the footprints of historical migrations in the Iberian Peninsula
The Iberian Peninsula has a complex history. Here, the authors analyse the genetic structure of the modern Iberian population at fine scale, revealing historical population movements associated with the time of Muslim rule.
- Clare Bycroft
- , Ceres Fernandez-Rozadilla
- & Simon Myers
-
Article
| Open Access Chromatin interactions and expression quantitative trait loci reveal genetic drivers of multimorbidities
Chromatin interactions and expression quantitative trait loci reveal genetic drivers of multimorbidities
Multimorbidities of common diseases often have shared underlying predisposing factors. Here Fadason et al. study the pleiotropy of SNPs and their effects on target genes by integrating chromatin interaction and expression quantitative trait loci data to identify target genes shared between phenotypes.
- Tayaza Fadason
- , William Schierding
- & Justin M. O’Sullivan
-
Article
| Open Access Phenome-wide association studies across large population cohorts support drug target validation
Phenome-wide association studies across large population cohorts support drug target validation
Testing the association between genetic variants and a range of phenotypes can assist drug development. Here, in a phenome-wide association study in up to 697,815 individuals, Diogo et al. identify genotype–phenotype associations predicting efficacy, alternative indications or adverse drug effects.
- Dorothée Diogo
- , Chao Tian
- & Heiko Runz
-
Article
| Open Access Germline pathogenic variants of 11 breast cancer genes in 7,051 Japanese patients and 11,241 controls
Germline pathogenic variants of 11 breast cancer genes in 7,051 Japanese patients and 11,241 controls
Association between variants in 11 different genes and breast cancer risk has been established and sequencing of these genes is recommended to provide personalized diagnosis, therapy, and surveillance for the high-risk patients and their relatives. Here the authors analyse the frequency of germline pathogenic mutations in these genes specifically in a Japanese population.
- Yukihide Momozawa
- , Yusuke Iwasaki
- & Michiaki Kubo
-
Article
| Open Access De novo human genome assemblies reveal spectrum of alternative haplotypes in diverse populations
De novo human genome assemblies reveal spectrum of alternative haplotypes in diverse populations
The majority of the human reference genome assembly is represented as a single consensus haplotype. Here, Wong et al. analyze de novo assemblies of 17 diverse, haplotype-resolved genomes to gain insights into the structure of genetic diversity and compile a list of alternative haplotypes across populations.
- Karen H. Y. Wong
- , Michal Levy-Sakin
- & Pui-Yan Kwok
-
Correspondence
| Open AccessReply to ‘Misestimation of heritability and prediction accuracy of male-pattern baldness’
- Nicola Pirastu
- , Peter K. Joshi
- & James F. Wilson
-
Article
| Open Access Analysis of predicted loss-of-function variants in UK Biobank identifies variants protective for disease
Analysis of predicted loss-of-function variants in UK Biobank identifies variants protective for disease
Examination of predicted loss-of-function (pLOF) genetic variants allows direct identification of genes with therapeutic potential. Here, Emdin et al. perform association analysis for 3759 pLOF variants with 24 traits and highlight protective variants against cardiometabolic and immune phenotypes.
- Connor A. Emdin
- , Amit V. Khera
- & Sekar Kathiresan
-
Article
| Open Access Medical relevance of protein-truncating variants across 337,205 individuals in the UK Biobank study
Medical relevance of protein-truncating variants across 337,205 individuals in the UK Biobank study
Protein-truncating variants (PTVs) are predicted to significantly affect a gene’s function and, thus, human traits. Here, DeBoever et al. systematically analyze PTVs in more than 300,000 individuals across 135 phenotypes and identify 27 associations between PTVs and medical conditions.
- Christopher DeBoever
- , Yosuke Tanigawa
- & Manuel A. Rivas
-
Article
| Open Access GWAS for male-pattern baldness identifies 71 susceptibility loci explaining 38% of the risk
GWAS for male-pattern baldness identifies 71 susceptibility loci explaining 38% of the risk
Male pattern baldness (MBP) is a complex trait that has genetic associations. Here, Pirastu and colleagues perform a genome-wide association study to show 71 susceptibility loci associated with MBP — 30 of which are novel — and that these loci can explain 38% of heritability.
- Nicola Pirastu
- , Peter K. Joshi
- & James F. Wilson
-
Article
| Open Access Prevalence of sexual dimorphism in mammalian phenotypic traits
Prevalence of sexual dimorphism in mammalian phenotypic traits
Systemic dissection of sexually dimorphic phenotypes in mice is lacking. Here, Karp and the International Mouse Phenotype Consortium show that approximately 10% of qualitative traits and 56% of quantitative traits in mice as measured in laboratory setting are sexually dimorphic.
- Natasha A. Karp
- , Jeremy Mason
- & Jacqueline K. White
-
Article
| Open Access Improved imputation of low-frequency and rare variants using the UK10K haplotype reference panel
Improved imputation of low-frequency and rare variants using the UK10K haplotype reference panel
Imputation uses genotype information from SNP arrays to infer the genotypes of missing markers. Here, the authors show that an imputation reference panel derived from whole-genome sequencing of 3,781 samples from the UK10K project improves the imputation accuracy and coverage of low frequency variants compared to existing methods.
- Jie Huang
- , Bryan Howie
- & Nicole Soranzo
-
Article |
 Enhanced meta-analysis and replication studies identify five new psoriasis susceptibility loci
Enhanced meta-analysis and replication studies identify five new psoriasis susceptibility loci
About 2% of the population are affected by psoriasis, a chronic skin disease with complex genetics. Here Tsoi et al.conduct a meta-analysis of several genome-wide association studies and identify five novel loci, helping to further our understanding of the biology behind this condition.
- Lam C. Tsoi
- , Sarah L. Spain
- & James T. Elder
-
Article |
 Human gephyrin is encompassed within giant functional noncoding yin–yang sequences
Human gephyrin is encompassed within giant functional noncoding yin–yang sequences
Yin–yang haplotypes are stretches of DNA that differ at multiple markers and exhibit two disparate forms. Here, the authors identify a pair of 284-nucleotide-long yin–yang haplotypes that encompass the gephyringene, and show that these human-specific haplotypes evolved rapidly and bear functional implications.
- Sharlee Climer
- , Alan R. Templeton
- & Weixiong Zhang
-
Article
| Open Access Unravelling the hidden ancestry of American admixed populations
Unravelling the hidden ancestry of American admixed populations
The genetic make-up of people living in the Americas has been shaped heavily by migration. Here the authors use a haplotype-based approach to reconstruct American genomic ancestry using genotype data from 2,500 individuals, revealing a previously unrecognized genetic contribution from European and African populations.
- Francesco Montinaro
- , George B.J. Busby
- & Cristian Capelli
-
Article
| Open Access Individual diet has sex-dependent effects on vertebrate gut microbiota
Individual diet has sex-dependent effects on vertebrate gut microbiota
Diet variations can alter gut microbial composition, but the potential influence of host genetic factors on these effects is unclear. Here, the authors show, in humans and in natural and laboratory fish populations, that such effects are dependent on the host’s sex, a genetically determined factor.
- Daniel I. Bolnick
- , Lisa K. Snowberg
- & Richard Svanbäck
-
Article |
 Integrating sequence and array data to create an improved 1000 Genomes Project haplotype reference panel
Integrating sequence and array data to create an improved 1000 Genomes Project haplotype reference panel
1000 Genomes imputation can increase the power of genome-wide association studies to detect genetic variants associated with human traits and diseases. Here, the authors develop a method to integrate and analyse low-coverage sequence data and SNP array data, and show that it improves imputation performance.
- Olivier Delaneau
- , Jonathan Marchini
- & Leena Peltonenz
-
Article |
 Neolithic mitochondrial haplogroup H genomes and the genetic origins of Europeans
Neolithic mitochondrial haplogroup H genomes and the genetic origins of Europeans
Here, Brotherton and colleagues sequence 39 mitochondrial genomes from ancient human remains. They track population changes across Central Europe and find that the foundations of the European mitochondrial DNA pool were formed during the Neolithic rather than the post-glacial period.
- Paul Brotherton
- , Wolfgang Haak
- & Janet S. Ziegle
-
Article
| Open Access Mouse urinary peptides provide a molecular basis for genotype discrimination by nasal sensory neurons
Mouse urinary peptides provide a molecular basis for genotype discrimination by nasal sensory neurons
Major histocompatibility complex peptide ligands in mouse urine have been hypothesized to serve as signals for communication. In support of this hypothesis, Sturm and colleagues find that specific urinary peptides from genetically different mouse strains can be discriminated by nasal sensory neurons.
- Theo Sturm
- , Trese Leinders-Zufall
- & Hans-Georg Rammensee
-
Article
| Open Access Gipc3 mutations associated with audiogenic seizures and sensorineural hearing loss in mouse and human
Gipc3 mutations associated with audiogenic seizures and sensorineural hearing loss in mouse and human
Progressive sensorineural hearing loss affects many people, but the underlying genetics remain largely undefined. Here, the authors identify mutations inGIPC3in mice and two consanguineous families that lead to hearing loss and in mice cause defects in the structure of stereocilia bundles and audiogenic seizures.
- Nikoletta Charizopoulou
- , Andrea Lelli
- & Konrad Noben-Trauth
 An ensemble penalized regression method for multi-ancestry polygenic risk prediction
An ensemble penalized regression method for multi-ancestry polygenic risk prediction