
Abstract
The 3-dimensional spatial and 2-dimensional frontal QRS-T angles are measures derived from the vectorcardiogram. They are independent risk predictors for arrhythmia, but the underlying biology is unknown. Using multi-ancestry genome-wide association studies we identify 61 (58 previously unreported) loci for the spatial QRS-T angle (N = 118,780) and 11 for the frontal QRS-T angle (N = 159,715). Seven out of the 61 spatial QRS-T angle loci have not been reported for other electrocardiographic measures. Enrichments are observed in pathways related to cardiac and vascular development, muscle contraction, and hypertrophy. Pairwise genome-wide association studies with classical ECG traits identify shared genetic influences with PR interval and QRS duration. Phenome-wide scanning indicate associations with atrial fibrillation, atrioventricular block and arterial embolism and genetically determined QRS-T angle measures are associated with fascicular and bundle branch block (and also atrioventricular block for the frontal QRS-T angle). We identify potential biology involved in the QRS-T angle and their genetic relationships with cardiovascular traits and diseases, may inform future research and risk prediction.
Similar content being viewed by others
Introduction
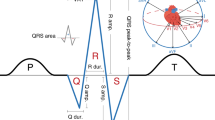
Abnormalities of ventricular depolarization and repolarization are a cause of malignant arrhythmia, which are associated with cardiac morbidity and mortality1. Mechanisms underlying the relationship of conventional electrocardiographic (ECG) measures with arrhythmogenesis (e.g. the QT interval and QRS duration) have previously been explored and highlight the role of cardiac ion channels. However, the biology reflected by markers derived from the vectorcardiogram is largely unknown2. These markers include the spatial (spQRSTa) and frontal (fQRSTa) QRS-T angles, which are the angles between the directions of ventricular depolarization and repolarization in 3- and 2-dimensional space, respectively (Fig. 1)3. Previous experimental and theoretical studies have shown that a wider QRS-T angle is determined through local variation in action potential duration and morphology4,5.

a Single lead electrocardiogram (ECG) signal with classical measures QRS duration and the QT interval labelled. The dark orange (estimates ventricular depolarization time) and blue (ventricular repolarization time) shaded sections of the signal represent the regions used to calculate the QRS and T-wave axes respectively with multiple ECG leads. b The spatial QRS-T angle (spQRSTa) mean is the angle between the mean amplitude of QRS and T-wave spatial loops. These spatial loops can be constructed from the resting 12-lead ECG using a standardised transformation, to produce representative X, Y and Z vectors that can be plotted over time. c The frontal QRS-T angle (fQRSTa) is the absolute difference between QRS and T-wave axes in the frontal plane only.
While vectorcardiographic measures are not currently used in routine clinical practice, there has been a resurgence of interest in their potential clinical utility, which has coincided with computational advances for efficient calculation of these markers. Recent studies have reported associations of the spQRSTa and fQRSTa with risk for arrhythmogenesis, sudden cardiac death and cardiac-related mortality6,7,8. In a population-based study, an abnormal spQRSTa was associated with a five-fold increased risk of cardiac and sudden death. No other conventional cardiovascular or ECG measure provided higher hazard ratios9. These measures may also be broad markers of cardiovascular risk, and associations have been reported with cardiomyopathies and cardioembolic stroke10,11. Improved knowledge of these markers will increase our understanding of these clinical relationships and has potential to identify new biology that is not captured by conventional ECG measures. Genome-wide association studies (GWAS) allow investigation of intermediate phenotypes and complex diseases to identify candidate genes and pathways that contribute to the underlying biology without a predefined hypothesis12. A previous GWAS meta-analysis for the spQRSTa (N = 13,826) identified 3 independent loci, with candidate genes involved in cardiac conduction and development13. However, this study was limited by a small sample size, and no GWAS has investigated the fQRSTa.
We performed the largest multi-ancestry studies to date for the spQRSTa (N = 118,780) and fQRSTa (N = 159,715) to identify additional candidate genes and pathways enriched for these markers, to advance our understanding of their genetic relationship with other ECG traits and cardiovascular disease, and to enhance the interpretation of existing and future clinical studies.
Results
Meta-analysis of QRS-T angle GWAS
Our primary multi-ancestry GWAS meta-analysis for spQRSTa had a total sample size of 118,780 individuals, including European (81.3%), Hispanic/Latino (10.7%) and African (7%) ancestries from 14 studies. The multi-ancestry GWAS meta-analysis for fQRSTa included 159,715 individuals from 23 studies and a similar ancestral composition (Supplementary Data 1–3, Supplementary Note 1). Ancestry-stratified analyses were also conducted. Due to the non-normal distribution of the traits, results are for the rank-based inverse normal transformed phenotype, with reference to corresponding effect sizes from the raw-phenotype analyses (degrees [°]) for clinical interpretation. No inflation of tests statistics was identified, but early deviation from the reference line was observed in Quantile-Quantile (Q-Q) plots for multi-ancestry and European-ancestry meta-analyses (driven by a locus on chromosome 17; Supplementary Figs. 1 and 2).
Genome-wide significant loci
In multi-ancestry meta-analyses, we identified a total of 61 (58 previously unreported) and 11 lead genome-wide significant (GWS; P < 5 × 10−8) variants at independent loci associated with spQRSTa and fQRSTa, respectively (Figs. 2 and 3, Supplementary Data 4 and 5). All lead variants for fQRSTa mapped within a locus reported for spQRSTa. All previously reported loci for spQRSTa (NFIA, HAND1 and TBX3) were GWS and were the most significant loci. A total of 51 and 9 GWS independent loci were identified in European ancestry meta-analyses for spQRSTa and fQRSTa, respectively. All loci were also GWS in the corresponding multi-ancestry analysis, except one fQRSTa locus (TTN; Supplementary Data 4 and 5).

Manahattan plot for the spatial QRS-T angle (spQRSTa) meta-analysis. Two-sided P-values are plotted on the -log10 scale (Y-axis). The red horizontal line indicates genome-wide significance (P < 5 × 10−8). Variants within the boundaries of loci previously reported for the spatial QRS-T angle are labelled with the candidate gene and colored blue. Variants at previously unreported loci are green.

Manahattan plot for the frontal QRS-T angle (fQRSTA) meta-analysis. Two-sided P-values are plotted on the -log10 scale (Y-axis). The red horizontal line indicates genome-wide significance (P < 5 × 10−8). Variants within the boundaries of loci previously reported for the spatial QRS-T angle are labelled with the candidate gene and colored blue. Variants at previously unreported loci are green.
Conditional analyses and heritability estimates in European ancestry individuals
To identify additional signals, Genome-wide Complex Trait Analysis (GCTA, v1.26.0)14 was performed using European ancestry UK Biobank (UKB) participant meta-analysis summary statistics from 33,960 individuals. The analyses identified conditionally independent variants at 4 loci for spQRSTa and at 2 loci for fQRSTa (Supplementary Data 6).
Common SNP-based heritability was estimated in the same set of UKB participants with BOLT-Restricted Maximum Likelihood (BOLT-REML, v2.3.2) software15. Heritabilities of spQRSTa and fQRSTa were 22.3% and 6.8%, respectively (standard error [SE] 1.0%). European ancestry lead and conditionally independent variants explained 4.0% and 0.5% of the variance of spQRSTa and fQRSTa, respectively. Therefore, these variants explain approximately 17.8% and 7.4% of the SNP-based heritability of spQRSTa and fQRSTa, respectively.
Follow-up of loci for the spatial QRS-T angle
Over 96% (59/61) of the spQRSTa lead multi-ancestry variants were common (minor allele frequency [MAF] > 0.05). Across all loci, the lead variant with the largest effect size was rs117526881, located upstream of MYH7 (effect size 3.7° per allele). At each locus, Variant Effect Predictor (VEP, Ensembl release 99) was used to identify potential functional consequences of lead variants and their proxies (r2 > 0.8)16. Missense variants were identified at 6 (9.8%) loci (Supplementary Data 7). SIFT or Polyphen-2 prediction tools identified variants that were likely to be deleterious at 2 loci (ADPRHL1 and KANSL1). The KANSL1 locus contained missense variants in strong LD with the lead SNP (r2 > 0.94) in multiple genes (KANSL1, SSPL2C, MAPT and LRRC37A2). The lead variant (or a proxy) of five loci had a Combined Annotation Dependent Depletion (CADD) score \(\ge\)20, and were therefore predicted to be among the most deleterious variants in the genome (i.e., in the top 1%; Supplementary Data 8). The low frequency missense variant rs41306688 (effect size −2.5° per allele) at the ADPRHL1 locus had the highest CADD score (26.7).
To identify variants associated with tissue-specific gene expression in cardiovascular tissues, data were extracted from the Genotype-Tissue Expression (GTEx, v8) project17. At 11 loci, the lead variant or a proxy was a significant cis- expression quantitative trait locus (eQTL) variant in cardiac (left ventricular [LV], right atrial appendage [RAA]) or vascular (coronary or aorta artery) tissue (Supplementary Data 9). At 5 loci, we identified support for pairwise colocalization (BACH [RAA], C1QTNF4 [LV, aorta artery], CDH13 [LV, RAA], LINC00964 [LV] and MTSS1 [LV, RAA], and PKDCC [LV]; posterior probability [PP] > 0.75).
To predict the effects of gene expression in LV, RAA and vascular tissue on our phenotypes, a transcriptome-wide association study (TWAS) was performed with S-PrediXcan software. The expression of 33 genes was significantly associated with the spQRSTa (Bonferroni corrected threshold; P < 3.1 × 10−6), 26 of which mapped within GWS loci, and 10 were significant in multiple tissues (Supplementary Data 10). Increased expression was associated with an increase in spQRSTa for 17 genes, whereas an inverse relationship was found for 15 genes (Supplementary Fig. 3). For TMEM198, increased expression in the aorta was associated with an increase in spQRSTa, but an inverse relationship was observed in LV tissue. All other genes with significant findings in multiple tissues had concordant directions of effect.
Non-coding variants may influence cardiac electrophysiology through effects on regulatory elements and chromatin folding. We used 40 kb and ~4 kb-resolution long-range chromatin interaction (Hi-C) datasets to identify potential target genes of regulatory variants18,19. Promoter interactions were identified at 17 (27.9%) multi-ancestry loci in LV or RV tissues (Supplementary Data 11a, b). GWAS Analysis of Regulatory and Functional Information Enrichment with LD correction (GARFIELD) was used to test for enrichment of variants at DNase 1 hypersensitivity sites in specific tissues using European ancestry summary statistics. The strongest enrichment was in fetal heart tissue (P < 7.5 × 10−36); however, additional tissues were identified, including fetal renal pelvis, adult heart and brain (Supplementary Fig. 4).
With single nucleus Assay for Transposase-Accessible Chromatin using sequencing (snATAC-seq) data, we tested for enrichment of non-coding variants at open chromatin regions, to identify cell-type specific functional effects in adult heart, by utilizing Chromatin Element Enrichment Ranking by Specificity (CHEERS)20,21. Significant enrichment was observed across all variants in atrial and ventricular cardiomyocytes (Supplementary Fig. 5).
Reconstituted gene-sets in Data-driven Expression-Prioritisation Integration for Complex Traits (DEPICT) software were used to prioritize potential candidate genes based on overlapping functional pathways22. Significant gene-set enrichment (false discovery rate [FDR] < 0.01) was observed in cardiac tissues (ventricle, atrial and atrial appendage) (Supplementary Data 12). Significantly enriched Gene-Ontology (GO) biological processes were extracted from DEPICT pathway analyses (Supplementary Data 13). Redundant GO terms were removed and the remaining processes clustered using the reduce and visualise Gene Ontology (REVIGO) web application23. This analysis identified clusters of biological processes involved in: cardiac development (including embryonic heart tube morphogenesis, muscle structure development, trabeculae formation and vasculogenesis); muscle cell differentiation and regulation of organ growth; actin filament-based movement; and cardiac contraction and hypertrophy (Fig. 4). Significant KEGG pathways were dilated, hypertrophic and arrhythmogenic right ventricular cardiomyopathies; cardiac muscle contraction; and arginine and proline metabolism. The top 10 enriched mouse phenotypes included dilated cardiac chambers; ventricular wall thickness (thick and thin); and abnormal cardiac development (Supplementary Data 13).

All significant (false discovery rate <0.01) multi-ancestry spatial QRS-T angle (spQRSTa) gene-ontology (GO) biological processes from Data-driven Expression-Prioritization Integration for Complex Traits (DEPICT) software were analyzed using the Reduce and Visualize Gene Ontology (REVIGO) web application to remove redundant terms and cluster related nodes. Highly similar GO terms are linked by edges where the line width indicates the degree of similarity. Within each cluster, the colour gradient represents differences in the DEPICT gene-set enrichment two-sided P-values, with lighter gradients reflecting smaller enrichment P-values (therefore more significant) compared with other nodes in the same cluster.
A summary of bioinformatic annotations for all spQRSTa multi-ancestry loci is provided in Supplementary Data 14. These findings have been supplemented with additional trait-relevant information from: Online Mendelian Inheritance in Man (OMIM)24; the International Mouse Phenotyping Consortium25 (IMP); the Human Protein Atlas26; and PubMed literature reviews for each candidate gene. We also performed lookups of each lead variant in the Open Targets Genetics ‘Locus to Gene’ machine learning gene-prioritization pipeline for further annotations (Supplementary Data 14)27.
We identified two independent loci in the Hispanic/Latino spQRSTa meta-analysis, including one locus that was not GWS in the multi-ancestry meta-analysis (lead variant rs112628278, multi-ancestry GWAS P = 0.01). rs112628278 (nearest gene VAV2) is a low frequency Hispanic/Latino variant (MAF = 0.011) and rare among European ancestry individuals (MAF = 0.0002, 1000 Genomes [1000 G] reference panel).
One unreported locus (FAM135B) identified in the African ancestry spQRSTa meta-analysis showed no evidence for association in the multi-ancestry meta-analysis (P > 0.05). The lead variant (rs28377209) has a higher MAF in African ancestry populations, compared with Europeans (0.19 vs 0.10).
Follow-up of loci for the frontal QRS-T angle
Three variants at two loci were significant eQTL variants (LV [SSXP10, RP11-632C17_A.1], coronary artery [GNAZ]), but there was no support for colocalization (Supplementary Data 9). Eight genes were significant in the TWAS, and overlapped with spQRSTa genes, except for two (CEP85L and MMP11) (Supplementary Data 10). Tissue-specific promoter interactions were identified for variants at two loci that were not reported for spQRSTa loci (lead variant rs10885011; FAM124A and DLEU7, rs5030613; BCR) (Supplementary Data 11a, b). An unreported locus identified in the African ancestry fQRSTa meta-analysis was not GWS in spQRSTa analyses. The gene nearest to the lead signal is CCDC60 (Coiled-Coil Domain Containing 60).
Genetic correlation and overlap of GWS loci with other ECG measures
LD Score Regression (LDSC) software was used to estimate genetic correlations (rg) of spQRSTa and fQRSTa with ECG markers of cardiac conduction (PR interval), ventricular depolarization (QRS duration) and repolarization (QT and JT intervals)28,29. There was a high positive genetic correlation between spQRSTa and fQRSTa (rg = 0.61). Weak positive correlations wereobserved with PR interval (rg = 0.12, P = 6 × 10−4 for spQRSTa; rg = 0.19, P = 2.2 × 10−5 for fQRSTa). However, no statistically significant correlation was observed with the other ECG traits (Supplementary Fig. 6).
We used additional approaches to interrogate genetic overlaps. First, lead variants reported for other resting ECG traits were extracted and overlap was reported if they mapped within spQRSTa locus boundaries (within r2 > 0.1 or ±500 kb from the lead spQRSTa variant). Despite the low genetic correlations observed genome-wide, 26 (42.6%), 27 (44.3%) and 26 (42.6%) lead multi-ancestry spQRSTa variants mapped to reported PR, QRS and HR loci, respectively (Supplementary Data 15). Fewer variants mapped to reported QT and JT loci (19 [31.1%] and 14 [23%], respectively) (Fig. 5). Of the 7 loci reported for the global electrical heterogeneity trait SAI QRST, 3 lead variants mapped within the boundaries of spQRSTa loci (SCN5A, MYBPC3 and NDRG4).

Venn diagram showing spatial QRS-T angle (spQRSTa) multi-ancestry loci where a lead variant reported for another electrocardiographic ECG measure maps within the locus boundaries. For this figure, ECG measures shown are PR interval (cardiac conduction), QRS duration (ventricular depolarization), QT and JT intervals (ventricular repolarization) and heart rate (HR). Overlap was declared if a lead variant for these ECG measures mapped to within ±500 kb or r2 > 0.1 of a lead variant at a spQRSTa locus. Some loci overlap with other ECG traits (not visualised here but presented in Supplementary Data 15). At seven spQRSTa loci, no overlap was observed with any ECG trait (blue circle bottom right).
Next, we performed a pairwise GWAS with GWAS-PW, which uses Bayesian bivariate methods to estimate the probability for each genomic region that a variant affects both traits tested30. Across all spQRSTa loci, there was evidence for shared genetic influences at 17 (27.9%), 20 (32.8%), 7 (11.5%), 14 (22.9%) and 12 (19.7%) loci involving PR, QRS, HR, QT and JT, respectively (PP > 0.9). Of the loci that shared effects with QT and JT, 8/14 (57.1%) and 6/12 (50%) loci, respectively, also influenced QRS duration (Supplementary Data 15). The smallest P-value for variants at the NOS1AP locus in the spQRSTa multi-ancestry meta-analysis was 7.3 × 10−5. NOS1AP is the locus consistently reported with the strongest QT and JT associations. We performed a sensitivity analysis in ~34,000 UKB individuals to determine whether inclusion of the QT interval as a covariate influenced our findings. Beta estimates and P-values were highly correlated (rho[ρ] = 0.99 and 0.96 respectively) across all variants comparing a GWAS with or without the QT interval as a covariate. Also, there was no substantial change in the minimum P-value of variants at the NOS1AP locus.
At 7 multi-ancestry spQRSTa loci, we observed no overlap with previously reported ECG loci. Candidate genes at these loci include AHNAK2, ALDH1A2, SGCG and TAOK2.
Pleiotropy of genetic variants with other phenotypes
We performed a phenome-wide association study (PheWAS) to identify associations of European ancestry lead and conditionally independent spQRSTa variants with 1301 clinical conditions in 395,758 unrelated individuals European-ancestry individuals. Data on clinical conditions were from hospital episode statistics. Significantly associated conditions included atrial fibrillation, bundle branch block (BBB), atrioventricular block (AVB), arterial embolism and thrombosis, and hypertension (Fig. 6). We also performed lookups of all multi-ancestry lead spQRSTa variants (and proxies) in Phenoscanner (v2), to determine if they appeared in GWAS reports for non-ECG phenotypes and diseases (Supplementary Data 16). Lead variants or proxies at 19 spQRSTa loci (31.1%) had reported associations with blood pressure, anthropometric traits, blood counts, or psychiatric features or disorders (P < 5 × 10−8).

X-axis: Lead variant (RsID [Chromosome: Position (hg19): Allele1: Allele2]) or conditionally independent variant from the spatial QRS-T angle (spQRSTa) European ancestry meta-analysis that had a significant association with a clinical phenotype in UK Biobank. Y-axis: Phenotype derived from hospital episode statistics, with colour coding for each major group (circulatory system; red, digestive system; green, neoplasms; yellow, respiratory; blue). Odds ratios (OR) are color coded according to decreasing (blue) or increasing (green) odds. 3:38587306:A:G was a conditionally independent variant at the SCN5A locus.
Association of genetically determined spQRSTa and fQRSTa with cardiovascular disease
Polygenic risk scores (PRSs) were used to explore associations between genetically determined spQRSTa and fQRSTa and relevant cardiovascular diseases. PRSs were calculated by summing the dosage of lead variants from the European-ancestry meta-analysis, weighted by the effect size estimates from the corresponding untransformed analysis. To obtain preliminary β estimates for the association of PRSs with the directly measured ECG trait, we performed a linear regression adjusting for age, sex, RR interval, BMI, height and 10 genetic principal components, in 33,960 unrelated individuals of European ancestry from UKB. These individuals were included in the GWAS meta-analysis, and therefore β estimates and CIs are biased. However, approximation is useful to aid interpretation of subsequent analyses. Associations observed for each PRS were (β [95% CI]): 5.4° (5.1–5.7) for spQRSTa; and 2.03° (1.8–2.3) for fQRSTa (per standard deviation [SD] increase in the PRS).
Subsequently, each PRS was tested for association with prevalent cases of cardiovascular disease in 395,758 unrelated European ancestry UKB participants who were not in the GWAS meta-analysis (adjusting for sex, age, and 10 genetic principal components). We used a Bonferroni corrected threshold to identify significant findings (0.05/number of conditions tested, P < 6.3 × 10−3). Genetically determined spQRSTa was associated with increased odds for fascicular or bundle branch block (odds ratio [OR] (95% CI) per SD: 1.10 [1.07–1.13]) (Supplementary Fig. 7, Supplementary Data 17). Association of a QRS PRS with fascicular or bundle branch block has been reported31. However inclusion of a QRS PRS as a covariate did not substantially change the point estimates (1.09 [1.06–1.13]), supporting an interpretation that the spQRSTa PRS contains independent risk information. There was suggestive evidence for an association with AV block but not at the Bonferroni corrected significance threshold (OR: 1.04 [1.01–1.06], P = 7.7 × 10−3). Genetically determined fQRSTa was significantly associated with fascicular block or bundle branch block (OR 1.05 [1.02–1.08]), and AV block (OR 1.04 [1.01–1.07]).
No evidence for a causal relationship between spQRSTa and cardiomyopathies
Because candidate genes and pathway analyses indicated potential involvement with cardiomyopathies, we performed Mendelian randomization (MR) studies to test for a causal relationship of genetically determined spQRSTa (as the exposure) with hypertrophic cardiomyopathy32 (HCM) and idiopathic dilated cardiomyopathy33 (DCM) (as outcomes). Lead variants from multi-ancestry and European spQRSTa meta-analyses were used as instrumental variables (IV). A relationship was suggested with HCM (multi-ancestry: OR 1.01 [1.00–1.02], P = 0.004; European: 1.01 [1.00–1.02], P = 0.009), using a fixed-effect inverse variance-weighted (IVW) model. However, the association was not supported in sensitivity analyses, including MR-Egger, weighted median and MR-PRESSO analyses (Supplementary Data 18). Similarly, no causal relationship was identified with either sarcomere positive or sarcomere negative HCM cases. There were no significant findings in spQRSTa-DCM MR analyses (Supplementary Data 19). Funnel, scatter, and forest plots for HCM and DCM analyses are presented in Supplementary Figs. 8 and 9.
Discussion
Our large-scale analyses of spQRSTa and fQRSTa -- two emerging markers for arrhythmogenesis and cardiovascular disease -- significantly advance our understanding of their basic biology and relationships with classical ECG markers. We identify candidate genes involved in cardiac development, muscle cell differentiation, cardiac contraction and actin-filament based movement. The genes also haverelationships with cardiomyopathies and central arterial vascular development. spQRSTa and fQRSTa shared loci with other ECG measures. But there are also 7 unshared loci, suggesting distinct genetic influences. Among spQRSTa and fQRSTa loci, there are fewer genes for cardiac ion channels, in contrast to findings for other ECG traits. Based on a phenome-wide scan, we report associations with atrial fibrillation, conduction disease and arterial embolism. Moreover, PRSs are associated with fascicular and bundle branch block, and AV block, indicating potential downstream effects of the loci.
A substantial proportion of lead candidate genes at spQRSTa loci are associated with development of inherited cardiomyopathies in humans (including MYH7, TTN, TNNT2, MYBPC3, DSP, RBM20; Fig. 7)34. There was also support for genes with non-Mendelian roles in cardiac myogenesis, including ADPRHL1, NACA and NFIA. The function of ADPRHL1 in humans has yet to be established, however, knockout of ADPRHL1 in Xenopus laevis causes loss of the myofibril assembly in ventricular cardiomyocytes and prevents ventricular outgrowth35.

Candidate genes at spatial QRS-T angle (spQRSTa) loci are grouped according to potential roles in embryonic development, cardiac structure and function. RYR2 and ACTN2 are candidate genes from the same locus. A summary of the bioinformatic evidence for each gene is presented in Supplementary Data 14. Created using BioRender.com.
Small clinical studies have identified an association between a widened spQRSTa and HCM in paediatric and adult populations10,36. A widened spQRSTa also predicts occurrence of ventricular arrhythmia among HCM patients37. Interestingly, we did not identify a causal relationship between genetically determined spQRSTa and HCM or DCM in MR studies. Lack of association could be due to the small sizes of the HCM and DCM cohorts. However, the analyses did identify GWS loci. Therefore, the spQRSTa may reflect functional information in these cardiomyopathies (conditional, non-obligatory), rather than causal mechanisms for the structural phenotype. The spQRSTa may also reflect mechanisms and conditions predisposing to intermittent changes in ventricular conduction (e.g., intermittent or persistent BBB) indicating the development of cardiac memory11,38. This is supported by our PheWAS and PRS analyses, where we observed associations with fascicular or bundle branch block and AV block. Therefore, although we did not find a causal relationship with structural HCM or DCM phenotypes, the spQRSTa may reflect the burden of intermittent ventricular arrhythmia or conduction abnormalities occurring over time in these conditions39.
Multiple findings support a role for angiogenesis and arterial development in modulating the spQRSTa, including candidate genes (ALDH1A2, ANGPT1, and VAV2), significant enrichment of GO-terms (coronary vascular development and vasculogenesis), and associations identified in PheWAS (arterial embolism, thrombosis and hypertension). VAV2, a candidate gene identified in Hispanic/Latino ancestry-specific analyses, is a guanine nucleotide exchange factor for Ras-related GTPases and modulates receptor-mediated angiogenic responses40,41. Knockout mice for this gene show signs of left ventricular hypertrophy, cardiac fibrosis and hypertension42. Abnormal angiogenesis influences cardiac structure and function through physiological and pathological cardiac hypertrophy, effects on tissue recovery following ischaemia, and regenerative capacity43,44. These processes may potentially lead to an arrhythmogenic substrate. A recent study identified an association between a widened spQRSTa and increased risk for cardioembolic and haemorrhagic stroke45. Our findings provide potential biological explanations for stroke associations.
Previous theoretical studies suggested that the spQRSTa reflects abnormalities of ventricular repolarization due to abnormal depolarization3. We identified shared genetic influences and loci overlapping with mainly PR interval and QRS duration. We also report loci that are shared across multiple ECG traits including NFIA, CASQ2, RYR2, TTN, SCN5A, PITX2, CDKN1A, PLN, NACA and NDRG4 (Fig. 7, Supplementary Data 15). In comparison to results reported for QT and JT, there is less support for the involvement of cardiac potassium channels, which are important determinants of ventricular repolarization and common targets of existing anti-arrhythmics46. Combined with other studies, our results support an interpretation that the spQRSTa is primarily a marker of abnormal ventricular depolarization and suggests new therapies targeting depolarization should be investigated for arrhythmia prevention and management.
Despite evidence for shared effects at some loci, genetic and phenotypic correlations of spQRSTa and fQRSTa with other ECG traits are weak. Therefore, spQRSTa and fQRSTa may represent unique biology that may contribute to arrhythmic risk. There was no overlap with other ECG traits at 7 multi-ancestry spQRSTa loci. Candidate genes at these loci include: AHNAK2, which encodes a large nucleoprotein that localises to the Z-band region of mouse cardiomyocytes and may have a role in excitation-contraction coupling through effects on L-type voltage-gated calcium channels; SGCG, a component of the subsarcolemmal cytoskeleton; and ALDH1A2, which encodes an enzyme responsible for early embryonic retinoic acid synthesis, a process that is critical for normal cardiac and arterial development47,48,49,50. Another candidate gene TAOK2, is a protein kinase most studied for its role in dendritic spine maturation51. More recently, TAOK2 has been identified in tethering the endoplasmic reticulum to microtubules. We report another locus, MACF1, that is also involved in microtubule organization52,53. Validation of these loci is required.
Although sample sizes were significantly larger for fQRSTa than for spQRSTa (134% larger), we found fewer loci and lower heritability estimates for fQRSTa. All multi-ancestry fQRSTa loci overlapped with spQRSTa loci. There were candidate genes involved in cardiac development and cardiomyopathies including SCN5A, RBM20, PLN, TBX3 and MYO18B. The fQRSTa represents the QRS-T angle in the frontal plane only, whereas the spQRSTa is 3-dimensional. Therefore the fQRSTa trait likely loses information that resides in other planes. However, we identified an unreported locus in African ancestry-specific analyses (candidate gene FAM135B). Knockdown of FAM135B in iPSC lines reduces spinal motor neuron survival and contributes to neurite defects as seen in spinal and bulbar muscular atrophy. These disorders are associated with cardiac arrhythmia and structural abnormalties54,55,56.
Although our study includes individuals from multiple ancestries, ancestry-specific analyses were limited by sample sizes. Larger studies are needed to yield additional signals. The precise algorithms used to calculate the spQRSTa will marginally differ despite efforts to harmonise approaches; however, such differences are unlikely to affect our positive findings (measurement error or noise will dampen signals), and summary statistics for spQRSTa across all studies are broadly similar (Supplementary Data 3)57,58,59.
In summary, our analyses significantly advance our knowledge of the underlying biology reflected by the spQRSTa and fQRSTa, which are independent risk markers for arrhythmogenesis. We also identified loci that have not been reported for ECG traits. Our findings highlight biological processes and candidate genes that may explain associations observed in previous clinical studies and could inform future research on the utility of these markers in risk prediction.
Methods
Study cohorts
Fourteen studies (32 ancestry-specific sub-studies) and 23 studies (40 ancestry-sub-studies) contributed GWAS summary statistics for spQRSTa and fQRSTa meta-analyses, respectively. These included members of the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) consortium60 (Supplementary Data 1). This study was approved by all participating cohorts. Ethics and consent was obtained at a study level. The majority of participating cohorts were population based with a small number of case-control studies. Information for study level genotyping method (typically Illumina or Affymetrix), quality control (Hardy-Weinberg equilibrium [HWE] P, and MAF), are provided in Supplementary Data 2. The 1000 G reference panel (26/40 sub-studies) was most used for imputation (26/40) followed by the Haplotype Reference Consortium panel (13/40). The Atherosclerosis Risk in Communities (ARIC) study was imputed with TOPMed Freeze 5 reference panel61,62. All GWAS summary data included in the meta-analyses utilized NCBI build 37 (summary statistics for ARIC sub-studies were converted from build 38 to 37 using a liftover tool [https://genome.sph.umich.edu/wiki/LiftOver]).
Cohort-level single variant association analyses
A GWAS was performed by each participating cohort for the spQRSTa (mean) and fQRSTa. If the spQRSTa was not already calculated and digitized ECGs were available, it was derived by transformation of the 12-lead ECG using previously published algorithms57. In brief, after applying a bandpass butterworth filter and signal averaging to reduce noise, orthogonal X, Y and Z vector beats were estimated using Kors’ regression matrix63. The spQRSTa was subsequently calculated as the angle between mean QRS and T-wave spatial vector loops57,58. The fQRSTa was defined as the absolute difference between QRS and T-wave frontal plane axes (fQRSTa = abs[QRS-axis − T-axis])3. Values for both phenotypes are between 0 and 180°.
The primary analysis for this study to declare GWS and previously unreported associations, was the rank-based inverse normal transformed phenotype (as both the spQRSTa and fQRSTa are not normally distributed). The raw phenotype was also analysed to calculate clinically meaningful effect sizes (on the degree [°] scale). Study level GWASs were performed using an additive genetic model, accounting for relatedness with appropriate software (e.g. BOLT linear mixed model [BOLT-LMM])15 or by including a kinship matrix or pedigree64,65,66. Poorly imputed genotypes were excluded (Rsq > 0.3 or similar for IMPUTE) and a MAF > 0.01 was applied, so that only high-quality variants were included in the study.
Summary statistics for cohort level distributions of each ECG trait and covariates included in the GWAS model, are provided in Supplementary Data 3. Age, sex, RR interval, height, and body-mass index (BMI) were mandatory covariates in the GWAS model. In addition, as the QT interval is associated with the QRS-T angle and we wished to identify associations that were not primarily driven by this marker of ventricular depolarization and repolarization, the QT interval was also included as a covariate. If pedigree data was not available, or if the chosen GWAS software did not correct for underlying population stratification, genetic principal components (PCs) were also included as covariates. Cohorts could also select additional covariates if relevant to their study, such as genotyping method or recruitment site. Cohorts comprising multiple ancestries performed separate analyses for each ancestry.
Individuals were not included in the study-level GWAS if they had a prior diagnosis of heart failure, myocardial infarction, pacemaker or implantable cardiac defibrillator; were prescribed class I or III anti-arrhythmics, QT-prolonging or digitalis medication; or were pregnant at the time of ECG acquisition. In addition, individuals were excluded if atrial fibrillation, BBB or a QRS duration greater than 120 ms, was present on their ECG.
Additional quality control of study-level data
After submissions of results in a standardized format, quality control was performed using EasyQC (R package v9.2)67. Allele frequencies of all variants were compared to those reported in the reference panel used by the study for imputation. To identify analytical errors, QQ and P–Z-score plots were inspected, and summary statistics for β estimates and SE were compared across all studies. To identify potential uncorrected population stratification, the genomic-control inflation factor was calculated to identify test statistic inflation.
GWAS meta-analysis
The primary GWAS meta-analysis for spQRSTa and fQRSTa was the multi-ancestry rank-based inverse normal transformed meta-analysis; however, to estimate clinically relevant effect sizes, a GWAS meta-analysis was also performed using the untransformed phenotype (on the degree [°] scale). European, African, and Hispanic/Latino ancestry-specific meta-analyses were also performed as secondary analyses. Meta-analyses were performed with METAL (v2011-03-25) using an IVW model68. If a study’s λ was >1.0, genomic control during the meta-analysis. Summary statistics and plots were produced for the entire meta-analysis. Subsequently in downstream analyses, variants were only included if present in >60% of the total meta-analysis sample size. The GWS threshold was set as P < 5 × 10−08. To calculate the correlation between variants, relevant individuals from the 1000 G reference panel were used; all individuals for the multi-ancestry summary statistics, ancestry-specific for European, African and Hispanic/Latino analyses. Some in-silico analyses relied upon correlations calculated by the software developers and did not permit modification. In these instances (and explicitly stated in the manuscript text), only European-ancestry summary statistics were used in recognition that the multi-ancestry meta-analysis contained a substantial proportion of individuals of European descent.
Definition of known and previously unreported loci
One previous GWAS has been reported for spQRSTa, with 3 loci reaching GWS13. Using PLINK (v1.9)69, lead variants from the study were extracted to calculate locus boundaries, defined as ±500 kb or r2 < 0.1 within a 4 mb region (whichever was larger), centered on the lead variant. The 1000 G reference panel was used to calculate correlations between variants61. The variants furthest upstream or downstream were declared the locus start and end respectively. We used the same list to define known loci for fQRSTa as no previous GWAS has been reported for this trait and as the phenotypic correlation with spQRSTa is high. The same method was used to identify GWS loci in our study. Loci that did not overlap with the list of known loci, were declared as previously unreported.
Heterogeneity I2 statistics and forest plots were produced for each lead variant (smallest P) at each locus, to identify evidence for heterogeneity. Locus-Zoom plots were also produced to visually inspect patterns of association and variant correlations70.
Conditional and heritability analyses
To identify independent secondary signals within GWS loci, conditional analyses using European-ancestry statistics were performed using Genome-wide Complex Trait Analysis (GCTA, v1.26.0)14. As recommended by GCTA, the largest cohort in the meta-analysis was used as the reference panel (UKB, N = 33,960). For this analysis, related individuals in the UKB sample were excluded (up to the 2nd-degree [kinship coefficient <0.0884]). A strict threshold (r2 < 0.1 with the lead variant and PJoint < 5 × 10−08) was used to declare “conditionally independent” signals within loci.
Heritability estimates were calculated using the same UKB individuals of European-ancestry included in the GWAS meta-analysis, using BOLT-REML (v2.3.2)15. BOLT-REML models directly genotyped SNPs to estimate relatedness within a sample and obtains SNP-based heritability estimates. The percentage of variance explained (PVE) by lead and conditionally independent variants was subsequently calculated (Eq. 1)71;
Variant annotation
Lead and conditionally independent variants (and their proxies [r2 > 0.8]) were annotated using Variant Effect Predictor (VEP, Ensembl release 99) to identify potential functional consequences16. VEP also contains data from prediction tools Sorting Intolerant From Tolerant algorithm (SIFT, version 5.2.2)72 and PolyPhen-2 (v2.2.273), which supplied deleteriousness scores. In addition, CADD74 and RegulomeDB75 scores for each of these variants were extracted. CADD scores annotate coding and non-coding variants, and enable ranking of their potential deleteriousness compared with other variants in the genome74.
Association with tissue-specific gene expression
To identify relationships between lead and conditionally independent variants (and their proxies), with tissue-specific gene expression, cis-eQTL data was extracted from the GTEx portal (v8)17,76,77. Tissues included in these analyses were cardiac (LV and RAA) and vascular (coronary and aorta artery), for their known influence on cardiovascular disease. If a variant was also a lead cis-eQTL variant, colocalization analysis were performed at the locus using the R package COLOC, to determine whether the variant was causal in both the GWAS meta-analysis and the eQTL study78. These colocalization analyses use Bayesian statistical methods to calculate a posterior probability (PP) for the variant being causal in both analyses (PP > 75%).
To predict the effects of gene expression levels on spQRSTa and fQRSTa, we performed a TWAS using S-PrediXcan. S-PrediXcan is an extension of the original software PrediXcan and infers results using GWAS summary statistics, thus removing the need for individual-level genotype and phenotype data79. S-PrediXcan provides a precalculated transcriptome model database from GTEX-based tissues and covariance matrices of SNPs within each gene model (https://github.com/hakyimlab/MetaXcan). We used European meta-analysis summary statistics for these analyses and tested for association in a total of 16,097 genes across LV, RAA and vascular tissues. A Bonferroni corrected threshold (0.05/number of genes tested [16,097] = 3.1 × 10−6) was used to declare significance and results are only reported when more than one SNP was included in the model.
Tissue- and cell-type specific regulatory elements
GARFIELD (v2) was used to identify tissue-specific enrichment of variants at DNase I hypersensitivity sites80. GARFIELD annotates variants with data from the ENCODE, GENCODE and Roadmap Epigenomics projects and calculated odd ratio using a generalised linear model framework80.
Chromatin interaction data was used to identify target genes of regulatory variants (RegulomeDB score ≤3b) in LV and RV tissues. First, using FUMA GWAS (Functional Mapping and Annotation of Genome-Wide Association Studies) software (v1.3.6), overlap was identified between lead and conditionally independent variant, and pre-processed loops determined by Fit-Hi-C pipelines18,81. An FDR threshold <0.05 was used to report results. In addition, we performed the same analysis using loops called from recently published Knight-Ruiz normalised 5 kb, 10 kb and 25 kb resolution promotor capture Hi-C data19.
To identify cardiac cell-type specific enrichment of non-coding variants, we utilized accessible chromatin information from snATAC-seq data, for atrial and ventricular cardiomyocyte, smooth muscle, endothelial, adipocyte, macrophage, fibroblast, lymphocyte and nervous cells21. Using PLINK, our GWAS meta-analysis summary statistics were partitioned into haplotype blocks centered on each lead variant (r2 > 0.1 within a 2 Mb radius). Peaks within the lowest decile of total read counts from the snATAC-seq data were removed using a SNP enrichment method CHEERS (version accessed 2020)20, followed by quantile normalization of the remaining peak counts20. Enrichment of variants (one-sided P) within the ATAC-seq peaks was estimated and a Bonferroni-corrected threshold (0.05/number of cell-types) used to report significant findings.
Candidate gene prioritisation and pathway enrichment
To identify additional candidate genes at each locus, DEPICT (v3) software was used, that prioritizes genes according to common functional pathways. DEPCT calculates a membership probability for each gene within 14,461 reconstituted gene-sets22. Additional analyses were performed using DEPICT to identify pathway enrichment of these genes using Gene-Ontology (GO), Kyoto Encyclopaedia of Genes and Genomes (KEGG), REACTOME and the Mouse genetics initiative (MGI) data. DEPICT also performs gene-set tissue enrichment analyses using annotations from human Affymetrix microarray probes. For all analyses, an FDR < 0.01 was used to identify significant results. To visualise GO biological processes from the DEPICT spQRSTa multi-ancestry meta-analysis output were analysed using the REVIGO web application to remove redundant terms and cluster related nodes23. They were subsequently visualised using Cytoscape (v3.8.2)82.
The output of all bioinformatic analyses were pooled and supplemented with trait relevant information from Online Mendelian Inheritance in Man (OMIM)24 and International Mouse Phenotyping Consortium25 (IMP, www.mousephenotype.org) databases, the Human Protein Atlas26 (www.proteinatlas.org) and a PubMed literature review (Supplementary Data 14). We also performed a look up of each lead variant in the Open Targets Genetics “Locus to Gene” machine learning pipeline, which uses supervised learning to weight evidence from different sources and prioritize genes at a locus27. This database does not include trait-specific information in the pipeline and therefore it is used to supplement the analyses performed for this work. For each locus, the candidate gene with the most support across all lines of evidence is indicated. We also included a second gene if there is support from multiple analyses.
LD score regression
LD score regression with LDSC (v1.0.1), was performed to calculate the genetic correlation of the spQRSTa and fQRSTa with other ECG traits including PR, QRS, JT and QT intervals28. LDSC (v1.0.1) uses pre-computed LD scores and therefore these analyses were performed with European ancestry summary statistics only. These LD scores are used as weights in the regression model29.
Overlap of spQRSTa loci with other resting ECG traits and association with clinical phenotypes
Lead variants previously reported for other ECG markers including P-wave duration, atrioventricular conduction (PR interval12), ventricular depolarization (QRS duration83 and QRS voltage84), ventricular repolarization (JT83, QT83 and Tp-Tend intervals85) and HR86 were tested for overlap with spQRSTa loci (definition of overlap; if previously reported lead variants were within ±500 kb or r2 > 0.1 of the lead spQRSTa variant). Summary statistics for each ECG trait were also extracted and pairwise-GWASs performed using Bayesian bivariate analyses as implemented in GWAS-PW30. GWAS-PW combines GWAS summary statistics using the variance of effect sizes at each SNP to estimate the probability that a given genomic region contains a variant that influences both traits or distinct associations, and learns reasonable thresholds from the data to declare significance. A pairwise GWAS was performed with the summary statistics of the multi-ancestry spQRSTa meta-analysis and each ECG trait of interest. To account for sample overlap between summary statistics, an expected correlation (-cor in GWAS-PW) between two traits was specified for each analysis87. The values used after adjusted for estimated sample overlap were; −0.0045, −0.0258, 0.0539, 0.0107, 0.0045 and 0.0135 for QT, JT, QRS, PR, HR and TpTe respectively. A posterior probability >0.9 was used as evidence supporting the presence of a causal SNP within the genomic region that influences both traits.
To identify evidence of pleiotropy with clinical conditions, a PheWAS was performed using the R package PheWAS (v0.99.5-5)88. ICD-10 and 9 codes were extracted from UKB hospital episode statistics and mapped to phecodes. Lead and conditionally independent variants from the European ancestry spQRSTa meta-analysis were subsequently tested for association with each phecode in 395,758 European individuals. Related pairs were excluded (kinship coefficient >0.0884). A Bonferroni corrected threshold for the number of phecodes tested (0.05/1,301 = 3.8 × 10−5) was used to declare significance. To identify evidence for pleiotropy with non-cardiac phenotypes and diseases from previously reported GWAS, a look-up was performed of lead and conditionally-independent spQRSTa variants (and proxies, r2 > 0.8) using Phenoscanner v289,90. Associations reaching GWS with other traits and diseases were extracted.
Sensitivity analyses
To determine whether the QT interval significantly influences the findings from our spQRSTa meta-analyses, sensitivity analyses were performed in UKB (N = 34,361). Analyses were repeated without the QT interval as a covariate. Spearman rank correlations (rho [ρ]) for beta estimates and -log10 P-values, were calculated across all variants between the original model and the sensitivity analysis.
Association between genetically determined spQRSTa and fQRSTa with cardiovascular diseases
A PRS was calculated for each trait using lead variants from the European meta-analysis, to test for association with atrial fibrillation, stroke, coronary artery disease, conduction disease, heart failure, non-ischaemic cardiomyopathy and ventricular arrhythmia. Analyses were performed in individuals of European ancestry in UKB (N = 395,758). Participants included in the GWAS meta-analysis were excluded, along with related pairs up to the 2nd-degree (kinship coefficient <0.0884). To take advantage of genotype probability data in BGEN format, PRSice-291 was used. The PRSs were calculated by summing the dosage of lead variants weighted by the effect size from the corresponding raw-phenotype meta-analysis. Disease status for each cardiovascular outcome of interest was extracted using ICD-9/ICD-10 codes from hospital admission episodes, self-reported data, operation codes and death certification (Supplementary Note 2). Associations were identified for prevalent and incident cases using a logistic regression model, including covariates age, sex, genotyping array and ten genetic principal components. A Bonferroni-corrected threshold of 0.05/number of outcomes tested (0.05/8 = 6.3.1 × 10−3) was used to declare significant associations.
Relationship between spQRSTa and HCM and DCM
The TwoSampleMR R package (v0.5.6), was performed to test for association of spQRSTa with cardiomyopathies, using data from cohorts with HCM and DCM92. First, summary statistics from a previously reported multi-ancestry (2780 cases, 47,486 controls) and European (2,244 cases, 42,668 controls) HCM GWAS were retrieved32. Summary statistics for multi-ancestry sarcomere positive (871 cases, 20,142 controls) and sarcomere negative (1874 cases, 27,344 controls) HCM GWAS were also extracted. The HCM GWAS included UKB participants as controls; however, there was no overlap of individuals included in the spQRSTa meta-analyses. Multi-ancestry (61 variants) and European (51 variants) IVs were constructed from GWS variants in the rank-based inverse normal transformed spQRSTa meta-analysis, with the corresponding β, SE and P retrieved from the untransformed meta-analysis to facilitate clinical interpretation. Effect alleles were harmonised between IVs and HCM summary statistics. Two variants, rs398110577 and rs35185344, from the multi-ancestry IV were unavailable within the HCM summary statistics and proxies were selected, rs4946230 (r2 = 0.70) and rs12928779 (r2 = 0.98), respectively. Four different methods were performed, specifically IVW, MR-Egger, weighted median and MR-PRESSO (mendelian randomisation pleiotropy residual sum and outlier), using MR-Base92,93. Results are reported as OR (95% CI) for risk of HCM per 1° increase in genetically determined spQRSTa. The same process was followed to test for association with DCM, but with the following differences. Summary statistics from a European ancestry “sporadic” DCM GWAS (2651 cases, 4329 controls) were used33. Sporadic DCM was defined as a reduced LV ejection fraction and enlarged LV end-diastolic volume/diameter in the absence of any obvious pathology33. For these analyses, one variant from the European IV was not available (rs2668692), therefore a suitable proxy was selected (rs10514897, r2 = 0.78).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Summary statistics from each genome-wide association study meta-analysis have been uploaded to the NHGRI-EBI Catalog of human genome-wide association studies website, https://www.ebi.ac.uk/gwas/.(Study accession numbers GCST90246318, GCST90246320, GCST90246322, GCST90246324 for Multi-ancestry, European, African and Hispanic ancestry meta-analyses for the spatial QRS-T angle, respectively. Study accession numbers GCST90246319, GCST90246321, GCST90246323, GCST90246325 for Multi-ancestry, European, African and Hispanic ancestry meta-analyses for the frontal QRS-T angle, respectively). Data relating to UK Biobank will be return to the study. The UK Biobank will make these data available to all bona fide researchers for all types of health-related research that is in the public interest, without preferential or exclusive access for any person. All researchers will be subject to the same application process and approval criteria as specified by the UK Biobank. Please see the UK Biobank’s website for the detailed access procedure (http:/www.ukbiobank.ac.uk/register-apply/). Other datasets used in these analyses are publicly available and can be sourced from: 1000 Genomes reference panel: https://www.internationalgenome.org/category/reference/; Haplotype reference consortium reference panel: http://www.haplotype-reference-consortium.org/; Variant level annotation from Variant Effect Predictor (VEP), Ensembl release 99: https://www.ensembl.org/info/docs/tools/vep/index.html; Variant level Combined Annotation Dependent Depletion scores from Combined Annotation Dependent Depletion (CADD, v1.4): https://cadd.gs.washington.edu/; Variant level tissue-specific gene expression from The GTEx portal (v8): https://gtexportal.org/home/; HiC data from the Functional Mapping and Annotation of Genome-Wide Association Studies (FUMA GWAS, v.1.3.6): https://fuma.ctglab.nl/; DNaseI hypersensivity site enrichment data from GWAS Analysis of Regulatory and Functional Information Enrichment with LD correction (GARFIELD, v2): https://www.ebi.ac.uk/birney-srv/GARFIELD/; Gene-set, biological pathways and tissue expression data from Data-driven Expression-Prioritization Integration for Complex Traits (DEPICT, v3): https://github.com/perslab/depict; Variant level RegulomeDB scores from RegulomeDB (v.2.0.3): https://regulomedb.org/regulome-search/; A compendium of promoter-centered long-range chromatin interactions in the human genome (Jung et al., 2019): https://doi.org/10.1038/s41588-019-0494-8; Cardiac cell type-specific gene regulatory programs and disease risk association (Hocker et al., 2021): DOI: 10.1126/sciadv.abf1444; Druggable genome dataset from Finan et al., 2017: DOI: 10.1126/scitranslmed.aag1166; g:Profiler (accessed May 2021): https://biit.cs.ut.ee/gprofiler/gost; Online Mendelian Inheritance in Man database: https://www.omim.org/; Mouse Genome Informatics: http://www.informatics.jax.org/; KEGG drug database: (https://www.genome.jp/).
Code availability
Codes are available from the original software used for each analysis.
References
Fishman, G. I. et al. Sudden cardiac death prediction and prevention: report from a national heart, lung, and blood institute and heart rhythm society workshop. Circulation 122, 2335–2348 (2010).
Joukar, S. A comparative review on heart ion channels, action potentials and electrocardiogram in rodents and human: extrapolation of experimental insights to clinic. Lab. Anim. Res. 37, 25 (2021).
Oehler, A., Feldman, T., Henrikson, C. A. & Tereshchenko, L. G. QRS-T angle: a review. Ann. Noninvasive Electrocardiol. 19, 534–542 (2014).
Plonsey, R. A contemporary view of the ventricular gradient of Wilson. J. Electrocardiol. 12, 337–341 (1979).
Young, W. et al. Comparisons of the spatial QRS-T angle with intra-cardiac markers of depolarization and repolarization. Comput. Cardiol. 2020 Computing in Cardiology, Rimini, Italy, pp. 1–4 (2020).
Zhang, X. et al. Spatial/frontal QRS-T angle predicts all-cause mortality and cardiac mortality: a meta-analysis. PLoS One 10, e0136174 (2015).
Jensen, K. et al. Bringing critical race praxis into the study of electrophysiological substrate of sudden cardiac death: the ARIC study. J. Am. Heart Assoc. 9, e015012 (2020).
Waks, J. W. et al. Competing risks in patients with primary prevention implantable cardioverter-defibrillators: Global Electrical Heterogeneity and Clinical Outcomes (GEHCO) study. Heart Rhythm. 18, 977–986 (2021).
Kardys, I. et al. Spatial QRS-T angle predicts cardiac death in a general population. Eur Heart J 24, 1357–1364 (2003).
Cortez, D., Schlegel, T. T., Ackerman, M. J. & Bos, J. M. ECG-derived spatial QRS-T angle is strongly associated with hypertrophic cardiomyopathy. J. Electrocardiol. 50, 195–202 (2017).
Johnson, J. A. et al. Electrophysiological ventricular substrate of stroke: a prospective cohort study in the Atherosclerosis Risk in Communities (ARIC) study. BMJ Open 11, e048542 (2021).
Ntalla, I. et al. Multi-ancestry GWAS of the electrocardiographic PR interval identifies 202 loci underlying cardiac conduction. Nat. Commun. 11, 2542 (2020).
Tereshchenko, L. G. et al. Genome-wide associations of global electrical heterogeneity ECG phenotype: The ARIC (atherosclerosis risk in communities) study and CHS (Cardiovascular Health Study). J. Am. Heart Assoc. 7, e008160 (2018).
Yang, J., Lee, S. H., Goddard, M. E. & Visscher, P. M. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82 (2011).
Loh, P. R. et al. Efficient Bayesian mixed-model analysis increases association power in large cohorts. Nat. Genet. 47, 284–290 (2015).
McLaren, W. et al. The ensembl variant effect predictor. Genome Biol. 17, 122 (2016).
Consortium, G. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660 (2015).
Schmitt, A. D. et al. A compendium of chromatin contact maps reveals spatially active regions in the human genome. Cell. Rep. 17, 2042–2059 (2016).
Jung, I. et al. A compendium of promoter-centered long-range chromatin interactions in the human genome. Nat. Genet. 51, 1442–1449 (2019).
Soskic, B. et al. Chromatin activity at GWAS loci identifies T cell states driving complex immune diseases. Nat. Genet. 51, 1486–1493 (2019).
Hocker, J. D. et al. Cardiac cell type-specific gene regulatory programs and disease risk association. Sci. Adv. 7, eabf1444 (2021).
Pers, T. H. et al. Biological interpretation of genome-wide association studies using predicted gene functions. Nat. Commun. 6, 5890 (2015).
Supek, F., Bošnjak, M., Škunca, N. & Šmuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One 6, e21800 (2011).
Online Mendelian Inheritance in Man, OMIM ®. McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University (Baltimore, MD), September 2022. World Wide Web URL: https://www.omim.org.
Dickinson, M. E. et al. High-throughput discovery of novel developmental phenotypes. Nature 537, 508–514 (2016).
Pontén, F., Jirström, K. & Uhlen, M. The human protein atlas–a tool for pathology. J. Pathol. 216, 387–393 (2008).
Carvalho-Silva, D. et al. Open Targets Platform: new developments and updates two years on. Nucleic Acids Res. 47, D1056–D1065 (2019).
Bulik-Sullivan, B. K. et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015).
Bulik-Sullivan, B. et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet. 47, 1236–1241 (2015).
Pickrell, J. K. et al. Detection and interpretation of shared genetic influences on 42 human traits. Nat. Genet. 48, 709–717 (2016).
Young, W. J. et al. Genetic analyses of the electrocardiographic QT interval and its components identify additional loci and pathways. Nat. Commun. 13, 5144 (2022).
Harper, A. R. et al. Common genetic variants and modifiable risk factors underpin hypertrophic cardiomyopathy susceptibility and expressivity. Nat. Genet. 53, 135–142 (2021).
Garnier, S. et al. Genome-wide association analysis in dilated cardiomyopathy reveals two new players in systolic heart failure on chromosomes 3p25.1 and 22q11.23. Eur. Heart J. 42, 2000–2011 (2021).
Muresan, I. D. & Agoston-Coldea, L. Phenotypes of hypertrophic cardiomyopathy: genetics, clinics, and modular imaging. Heart Fail Rev. 26, 1023–1036 (2020).
Smith, S. J., Towers, N., Demetriou, K. & Mohun, T. J. Defective heart chamber growth and myofibrillogenesis after knockout of adprhl1 gene function by targeted disruption of the ancestral catalytic active site. PLoS One 15, e0235433 (2020).
Cortez, D. et al. The spatial QRS-T angle outperforms the Italian and Seattle ECG-based criteria for detection of hypertrophic cardiomyopathy in pediatric patients. J. Electrocardiol. 48, 826–833 (2015).
Cortez, D., Graw, S. & Mestroni, L. In hypertrophic cardiomyopathy, the spatial peaks QRS-T angle identifies those with sustained ventricular arrhythmias. Clin. Cardiol. 39, 459–463 (2016).
Haq, K. T., Cao, J. & Tereshchenko, L. G. Characteristics of cardiac memory in patients with implanted cardioverter-defibrillators: the cardiac memory with implantable cardioverter-defibrillator (CAMI) study. J. Innov. Card Rhythm. Manag. 12, 4395–4408 (2021).
Perez-Alday, E. A. et al. Dynamic predictive accuracy of electrocardiographic biomarkers of sudden cardiac death within a survival framework: the Atherosclerosis Risk in Communities (ARIC) study. BMC Cardiovasc. Disord. 19, 255 (2019).
Tamás, P. et al. Mechanism of epidermal growth factor regulation of Vav2, a guanine nucleotide exchange factor for Rac. J. Biol. Chem. 278, 5163–5171 (2003).
Hunter, S. G. et al. Essential role of Vav family guanine nucleotide exchange factors in EphA receptor-mediated angiogenesis. Mol. Cell. Biol. 26, 4830–4842 (2006).
Sauzeau, V., Jerkic, M., López-Novoa, J. M. & Bustelo, X. R. Loss of Vav2 proto-oncogene causes tachycardia and cardiovascular disease in mice. Mol. Biol. Cell. 18, 943–952 (2007).
Lähteenvuo, J. & Rosenzweig, A. Effects of aging on angiogenesis. Circ. Res. 110, 1252–1264 (2012).
Cochain, C., Channon, K. M. & Silvestre, J. S. Angiogenesis in the infarcted myocardium. Antioxid. Redox Signal. 18, 1100–1113 (2013).
Johnson, J. A. et al. Does ventricular substrate play a role in incident stroke? The Atherosclerosis Risk in Communities (ARIC) study. medRxiv, https://doi.org/10.1101/2020.05.04.20090910 (2020).
Bihlmeyer, N. A. et al. ExomeChip-wide analysis of 95 626 individuals identifies 10 novel loci associated with QT and JT intervals. Circ. Genom. Precis. Med. 11, e001758 (2018).
Gardner, B. B., Swaggart, K. A., Kim, G., Watson, S. & McNally, E. M. Cardiac function in muscular dystrophy associates with abdominal muscle pathology. J. Neuromuscul. Dis. 2, 39–49 (2015).
Komuro, A. et al. The AHNAKs are a class of giant propeller-like proteins that associate with calcium channel proteins of cardiomyocytes and other cells. Proc. Natl. Acad. Sci. USA. 101, 4053–4058 (2004).
Niederreither, K. et al. The regional pattern of retinoic acid synthesis by RALDH2 is essential for the development of posterior pharyngeal arches and the enteric nervous system. Development 130, 2525–2534 (2003).
Moss, J. B. et al. Dynamic patterns of retinoic acid synthesis and response in the developing mammalian heart. Dev. Biol. 199, 55–71 (1998).
Yadav, S. et al. TAOK2 kinase mediates PSD95 stability and dendritic spine maturation through Septin7 phosphorylation. Neuron 93, 379–393 (2017).
Nourbakhsh, K., Ferreccio, A. A., Bernard, M. J. & Yadav, S. TAOK2 is an ER-localized kinase that catalyzes the dynamic tethering of ER to microtubules. Dev. Cell. 56, 3321–3333.e5 (2021).
Kakinuma, T., Ichikawa, H., Tsukada, Y., Nakamura, T. & Toh, B. H. Interaction between p230 and MACF1 is associated with transport of a glycosyl phosphatidyl inositol-anchored protein from the Golgi to the cell periphery. Exp. Cell. Res. 298, 388–398 (2004).
Sheila, M. et al. Phenotypic and molecular features underlying neurodegeneration of motor neurons derived from spinal and bulbar muscular atrophy patients. Neurobiol. Dis. 124, 1–13 (2019).
Wijngaarde, C. A. et al. Cardiac pathology in spinal muscular atrophy: a systematic review. Orphanet. J. Rare Dis. 12, 67 (2017).
Hattori, T., Ikeda, S., Yoshida, K., Yanagisawa, N. & Furihata, K. A patient with Kennedy-Alter-Sung syndrome showing cardiomyopathy. Rinsho Shinkeigaku 35, 1246–1249 (1995).
Young, W. J. et al. A method to minimise the impact of ECG marker inaccuracies on the spatial QRS-T angle: evaluation on 1,512 manually annotated ECGs. Biomed. Signal Process Control 64, 102305 (2021).
Perez-Alday, E. A. et al. Importance of the heart vector origin point definition for an ECG analysis: The Atherosclerosis Risk in Communities (ARIC) study. Comput. Biol. Med. 104, 127–138 (2019).
Haq, K. T. et al. Reproducibility of global electrical heterogeneity measurements on 12-lead ECG: the multi-ethnic study of atherosclerosis. medRxiv, https://doi.org/10.1016/j.jelectrocard.2021.09.014 (2021).
Psaty, B. M. et al. Cohorts for heart and aging research in genomic epidemiology (CHARGE) consortium: design of prospective meta-analyses of genome-wide association studies from 5 cohorts. Circ. Cardiovasc. Genet. 2, 73–80 (2009).
Auton, A. et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
McCarthy, S. et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet. 48, 1279–1283 (2016).
Kors, J. A., van Herpen, G., Sittig, A. C. & van Bemmel, J. H. Reconstruction of the Frank vectorcardiogram from standard electrocardiographic leads: diagnostic comparison of different methods. Eur. Heart J. 11, 1083–1092 (1990).
JR, O.C. MMAP User Guide. Available: http://edn.som.umaryland.edu/mmap/index.php. Accessed 7th December 2020.
Zhou, W. et al. Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat. Genet. 50, 1335–1341 (2018).
Kang, H. M. et al. Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 42, 348–354 (2010).
Winkler, T. W. et al. Quality control and conduct of genome-wide association meta-analyses. Nat. Protoc. 9, 1192–1212 (2014).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Pruim, R. J. et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 26, 2336–2337 (2010).
Shim, H. et al. A multivariate genome-wide association analysis of 10 LDL subfractions, and their response to statin treatment, in 1868 Caucasians. PLoS One 10, e0120758 (2015).
Kumar, P., Henikoff, S. & Ng, P. C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 4, 1073–1081 (2009).
Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 (2010).
Kircher, M. et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310–315 (2014).
Boyle, A. P. et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 22, 1790–1797 (2012).
Battle, A. et al. Genetic effects on gene expression across human tissues. Nature 550, 204–213 (2017).
Consortium, G. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 (2020).
Giambartolomei, C. et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet 10, e1004383 (2014).
Barbeira, A. N. et al. Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat. Commun. 9, 1825 (2018).
Iotchkova, V. et al. GARFIELD classifies disease-relevant genomic features through integration of functional annotations with association signals. Nat. Genet. 51, 343–353 (2019).
Watanabe, K., Taskesen, E., van Bochoven, A. & Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 8, 1826 (2017).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003).
Young, W. J. et al. Genetic analyses of the QT interval and its components in over 250K individuals identifies new loci and pathways affecting ventricular depolarization and repolarization. medRxiv, https://doi.org/10.1101/2021.11.04.21265866 (2021).
van der Harst, P. et al. 52 genetic loci influencing myocardial mass. J. Am. Coll. Cardiol. 68, 1435–1448 (2016).
Ramírez, J. et al. Common genetic variants modulate the electrocardiographic Tpeak-to-Tend interval. Am. J. Hum. Genet. 106, 764–778 (2020).
Mensah-Kane, J. et al. No clinically relevant effect of heart rate increase and heart rate recovery during exercise on cardiovascular disease: a mendelian randomization analysis. Front. Genet. 12, 569323 (2021).
Wang, J. et al. CAUSALdb: a database for disease/trait causal variants identified using summary statistics of genome-wide association studies. Nucleic Acids Res. 48, D807–D816 (2020).
Denny, J. C. et al. PheWAS: demonstrating the feasibility of a phenome-wide scan to discover gene-disease associations. Bioinformatics 26, 1205–1210 (2010).
Staley, J. R. et al. PhenoScanner: a database of human genotype-phenotype associations. Bioinformatics 32, 3207–3209 (2016).
Kamat, M. A. et al. PhenoScanner V2: an expanded tool for searching human genotype-phenotype associations. Bioinformatics 35, 4851–4853 (2019).
Choi, S. W. & O’Reilly, P. F. PRSice-2: polygenic risk score software for biobank-scale data. Gigascience 8, giz082 (2019).
Hemani, G. et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife 7, e34408 (2018).
Verbanck, M., Chen, C. Y., Neale, B. & Do, R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat. Genet. 50, 693–698 (2018).
Acknowledgements
All study acknowledgements can be found in Supplementary Note 3, and study funding information in Supplementary Note 4. W.J.Y acknowledges support by an MRC grant MR/R017468/1. A.A is supported by an NHLBI award K24HL148521. A.L.P.R is supported in part by CNPq (310679/2016-8 and 465518/2014-1) and by FAPEMIG (PPM-00428-17 and RED-00081-16). E.T-S is supported by Brazilian Ministry of Health (National Program of Genomics and Precision Health), Brazilian Conselho Nacional de Desenvolvimento Científico e Tecnologico (CNPq), Fundação de Amparo a Pesquisa do Estado de Minas Gerais (FAPEMIG, RED00314-16). M.F.L-C is supported by Brazilian Ministry of Health (DECIT/MS, EPIGEN-Brazil Project), Brazilian Ministry of Science and Technology (Financiadora de Estudos e Projetos (FINEP), Brazilian Conselho Nacional de Desenvolvimento Científico e Tecnologico (CNPq), Fundação de Amparo a Pesquisa do Estado de Minas Gerais (FAPEMIG)). M.L.S is supported by Brazilian Conselho Nacional de Desenvolvimento Científico e Tecnologico (CNPq), Third- World Academy of Science (TWAS), Fogarty International Center of the US National Institutes of Health (D43 TW007393). P.B.M, A.T, P.D.L and M.O acknowledge support by an MRC grant MR/N025083/1. P.B.M, H.R.W, A.T and P.D.L acknowledge the NIHR Barts Biomedical Research Centre at Queen Mary University of London. P.D.L is also supported by UCL/UCLH Biomedicine NIHR, Barts Heart Centre Biomedical Research Centre. J.R acknowledges support from the European Union’s Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement No.786833, and the “María Zambrano” fellowship through the European Union-NextGenerationEU. N.S is supported by grants AHA19SFRN348300063, R01HL141989, Medic One Foundation, Laughlin Family. U.S received funding from the Netherlands Heart Foundation (CVON2014-09, RACE V Reappraisal of Atrial Fibrillation: Interaction between hyperCoagulability, Electrical remodeling, and Vascular Destabilization in the Progression of AF), the European Union (ITN Network Personalize AF: Personalized Therapies for Atrial Fibrillation: a translational network, grant number 860974; MAESTRIA: Machine Learning Artificial Intelligence Early Detection Stroke Atrial Fibrillation, grant number 965286). S.T is supported by a Junior 1 Clinical Research Scholar award from the Fonds de Recherche du Québec-Santé (FRQS). J.L.I is supported by CACHET. M.O was supported by The John and Birthe Meyer Foundation and The Hallas-Møller Emerging Investigator Novo Nordisk (NNF17OC0031204). C.H is supported by an MRC University Unit Programme Grant MC_UU_00007/10 (QTL in Health and Disease). C.A and A.B were supported by NIH grants R01HL142825, and U01HG007416. D.D was supported by NIH grants R01HL138737 and T32HL139439. N.G, C.N and T.H are supported by the Novo Nordisk Foundation (Grant number NNF18CC0034900). H.M is supported by the CHARGE infrastructure grant (HL105756). J-W.B is funded by the Research Project CVON-AI (2018B017) financed by the PPP Allowance made available by Top Sector Life Sciences & Health to the Dutch Heart Foundation to stimulate public-private partnerships. D.M-K is supported by Dutch Science Organization (ZonMW-VENI Grant 916.14.023). J.F.W acknowledges support from the MRC Human Genetics Unit programme grant, “Quantitative traits in health and disease” (U. MC_UU_00007/10). L.R is funded by a University of Edinburgh studentship. P.N is supported by the MRC Human Genetics Unit programme grant, “Quantitative traits in health and disease” (U. MC_UU_00007/10). X.S was in receipt of a Starting Grant (2017-02543) from the Swedish Research Council (Vetenskaprådet). This research has been conducted using the UK Biobank Resource under Application Number 8256. This research used data assets made available by National Safe Haven as part of the Data and Connectivity National Core Study, led by Health Data Research UK in partnership with the Office for National Statistics and funded by UK Research and Innovation (grant ref MC_PC_20029). Copyright © (2022), NHS Digital. Re-used with the permission of the NHS Digital [and/or UK Biobank]. All rights reserved. The authors also wish to acknowledge the CHARGE infrastructure grant (HL105756).
Author information
Authors and Affiliations
Contributions
Interpreted results, writing, and editing the manuscript: W.J.Y, L.T, P.B.M. Conceptualization of project: W.J.Y, N.S, L.T, P.B.M. Supervision of project: L.T, P.B.M. Contributed to GWAS analysis plan: W.J.Y, N.S, L.T, P.B.M. Performed meta-analyses: W.J.Y and J.Ra. Performed GCTA, genetic correlations, heritability, variant annotations, GTEx analyses, HiC analyses, gene-set enrichment and pathway analyses, PheWAS, TWAS, Pairwise GWAS, Gene literature review, Phenoscanner look up, Comparison with existing ECG loci, PRS analyses in UKB: W.J.Y. Performed MR analyses using HCM GWAS meta-analysis summary statistics and provided text for the relevant section: A.R.H, A.G, C.G, H.Wat. Contributed data for DCM-spQRSTa MR analysis: S.G, J.-F.D, E.V, P.C. Provided support for bioinformatic analyses: B.M. Performed heritability analyses in ERF: A.I. Contributed to study-specific GWAS by providing phenotype, genotype and performing data analyses: W.J.Y, J.H, J.-W.B, L.Re, J.Y, A.I, J.Ra, S.V.D, A.R.B, M.P.C, T.D, L.F, J.L.I, H.M, R.N, C.N, A.Ric, M.Sa, C.M.S, N.Sor, S.Th, S.Tr, S.A, F.A, A.A, J.A.B, A.Ca, A.Co, D.D, A.D.L, C.E, C.F, X.G, T.H, S.R.H, R.D.J, J.A.K, M.F.L-C, A.L, P.W.M, A.C.M, P.N, D.J.P, P.P.P, A.Rei, L.Ri, U.S, X.S, G.S, E.Z.S, M.St, E.T-S, A.T, K.T, H.War, E.A.W, K.L.W, D.E.A, C.L.A, D.C, G.G, N.G, C.H, J.W.J, D.O.M-K, M.Ol, S.P, B.M.P, C.P, A.Rib, J.Ro, B.S, P.H, C.M.D, N.V, J.G.W, M.Or, C.K, H.J.L, J.F.W, J.K.K, N.Sot, P.D.L, P.B.M. All authors read, revised, and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
B.M.P serves on the Steering Committee of the Yale Open Data Access Project funded by Johnson & Johnson. D.C has received speaker fees from BMS/Pfizer and Servier, and consultation fees from Roche Diagnostics and Trimedics. U.S received consultancy fees or honoraria from Università della Svizzera Italiana (USI, Switzerland), Roche Diagnostics (Switzerland), EP Solutions Inc. (Switzerland), Johnson & Johnson Medical Limited, (United Kingdom), Bayer Healthcare (Germany). D.O.M.-K is a part time research consultant at Metabolon, Inc. U.S is co-founder and shareholder of YourRhythmics BV, a spin-off company of the University Maastricht. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Eric B. Fauman, Norihiro Kato and Roddy Walsh for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Young, W.J., Haessler, J., Benjamins, JW. et al. Genetic architecture of spatial electrical biomarkers for cardiac arrhythmia and relationship with cardiovascular disease. Nat Commun 14, 1411 (2023). https://doi.org/10.1038/s41467-023-36997-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-36997-w
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
