


















« Prev Next »
Mendel's studies of inheritance patterns in pea plants are a solid foundation for our current understanding of single-gene diseases in humans. Also called Mendelian or monogenic diseases, these kinds of diseases are caused by mutations in one gene, and they sometimes run in families. Pedigree analyses of large families with many affected individuals can be used to determine whether a disease-associated gene is located on an autosome or on a sex chromosome, and whether the related disease phenotype is dominant or recessive.
Autosomal Recessive Single-Gene Diseases
Autosomal recessive single-gene diseases occur only in individuals with two mutant alleles of the disease-associated gene. Remember, for any given gene, a person inherits one allele from his or her mother and one allele from his or her father. Therefore, individuals with an autosomal recessive single-gene disease inherit one mutant allele of the disease-associated gene from each of their parents. In pedigrees of families with multiple affected generations, autosomal recessive single-gene diseases often show a clear pattern in which the disease "skips" one or more generations.
Phenylketonuria (PKU) is a prominent example of a single-gene disease with an autosomal recessive inheritance pattern. PKU is associated with mutations in the gene that encodes the enzyme phenylalanine hydroxylase (PAH); when a person has these mutations, he or she cannot properly manufacture PAH, so he or she is subsequently unable to break down the amino acid phenylalanine, which is an essential building block of dietary proteins. As a result, individuals with PKU accumulate high levels of phenylalanine in their urine and blood, and this buildup eventually causes mental retardation and behavioral abnormalities.
The PKU-associated enzyme deficiency was determined biochemically in the 1950s—long before the PAH-encoding gene was mapped to human chromosome 12 and cloned in 1983. Specifically, Dr. Willard Centerwall, whose child was mentally handicapped, developed the first diagnostic test for PKU in 1957. Called the "wet diaper" test, Centerwall's test involved adding a drop of ferric chloride to a wet diaper; if the diaper turned green, the infant was diagnosed with PKU. The wet diaper test was used to reliably test infants at eight weeks after birth; by this time, however, infants who were affected by PKU had already often suffered irreversible brain damage.
Thus, in 1960, Dr. Robert Guthrie, whose niece suffered from PKU and whose son was also mentally handicapped, established a more sensitive method for detecting elevated phenylalanine levels in blood, which permitted a diagnosis of PKU within three days after birth. Guthrie's test used bacteria that were unable to make their own phenylalanine as messengers to report high blood levels of phenylalanine in an infant's blood sample obtained via heel prick. With Guthrie's method, the phenylalanine-deficient bacteria were grown in media together with a paper disk spotted with a drop of the infant's blood. If the phenylalanine levels in the blood were high, the bacteria would grow robustly, and a diagnosis of PKU could be made. Through the ability to discover that their child had PKU at such an early age, parents became able to respond immediately by feeding their child a modified diet low in proteins and phenylalanine, thereby allowing more normal cognitive development. Guthrie's test continues to be used today, and the practice of obtaining an infant's blood sample via heel prick is now used in numerous additional diagnostic tests.
Several other human diseases, including cystic fibrosis, sickle-cell anemia, and oculocutaneous albinism, also exhibit an autosomal recessive inheritance pattern. Cystic fibrosis is associated with recessive mutations in the CFTR gene, whereas sickle-cell anemia is associated with recessive mutations in the beta hemoglobin (HBB) gene. Interestingly, although individuals homozygous for the mutant HBB gene suffer from sickle-cell anemia, heterozygous carriers are resistant to malaria. This fact explains the higher frequency of sickle-cell anemia in today's African Americans, who are descendants of a group that had an advantage against endemic malaria if they carried HBB mutations. Finally, oculocutaneous albinism is associated with autosomal recessive mutations in the OCA2 gene. This gene is involved in biosynthesis of the pigment melanin, which gives color to a person's hair, skin, and eyes.
Autosomal Dominant Single-Gene Diseases
Autosomal dominant single-gene diseases occur in individuals who have a single mutant copy of the disease-associated gene. In this case, the presence of a single nonmutant or "wild-type" copy of the gene is not enough to prevent the disease. Individuals can inherit the mutant copy of the disease-associated gene from either an affected mother or an affected father.
Huntington's disease, a progressive neurodegenerative disorder, is a well-known example of an autosomal dominant single-gene disease; most individuals with a single copy of the mutant huntingtin gene (HTT) will have Huntington's disease later in life. Typically, autosomal dominant diseases affect individuals in their early years and prevent them from living past infancy or childhood, which in turn precludes these individuals from reproducing and potentially passing on the mutation to their offspring. In the case of Huntington's disease, however, the late onset of the disorder means that many affected individuals have already had children before they are even aware that they carry the mutation.
Disease-associated changes in the huntingtin gene consist of a special type of mutation called triplet repeats; these mutations are simply extra repetitions of the three-base DNA sequence CAG. The number of CAG repeats in a mutated huntingtin gene determines the age at which a person will develop Huntington's disease, as well as how severe the condition will be. Genetic tests can be used to determine how many CAG repeats are in an individual's huntingtin gene, thereby providing a highly accurate assessment of the individual's disease risk. Because affected parents have a 50% chance of passing a mutant copy of the huntingtin gene on to each of their offspring, children of people with Huntington's disease are often faced with the dilemma of whether to undergo such testing. Genetic testing can either provide immediate relief in knowing that one is free from the disease, or the confirmation that one will certainly suffer from the condition at some point in the future.
Myotonic dystrophy, familial hypercholesterolemia, neurofibromatosis, and polycystic kidney disease serve as additional examples of autosomal dominant single-gene diseases. Myotonic dystrophy is associated with dominant mutations in the dystrophia myotonica protein kinase (DMPK) gene; familial hypercholesterolemia is associated with dominant mutations in both the low-density lipoprotein receptor (LDLR) gene and the apolipoprotein B (APOB) gene; and neurofibromatosis is associated with dominant mutations in the neurofibromin (NF1) gene. Autosomal dominant polycystic kidney disease can be caused by mutations in either the polycystic kidney disease 1 (PKD1) gene or the polycystic kidney disease 2 (PKD2) gene; the PKD1 gene is located on human chromosome 16, whereas the PKD2 gene is located on human chromosome 4.
Table 1. Examples of Several Human Diseases, Their Modes of Inheritance, and the Associated Genes
| Type of Inheritance | Example | Gene Responsible |
| Autosomal recessive | Phenylketonuria | Phenylalanine hydroxylase (PAH) |
| Cystic fibrosis | Cystic fibrosis conductance regulator (CFTR) | |
| Sickle-cell anemia | Beta hemoglobin (HBB) | |
| Oculocutaneous albinism | OCA2 | |
| Autosomal dominant | Huntington's disease | Huntingtin (HTT) |
X Chromosome–Linked Recessive Single-Gene Diseases
Single-gene diseases that involve genes found on the sex chromosomes have somewhat different inheritance patterns than those that involve genes found on a person's autosomes. The reason for these differences lies in the genetic distinction between males and females. Recall that females have two copies of the X chromosome, and they receive one copy from each parent. Therefore, females with an X chromosome-linked recessive disease inherit one copy of the mutant gene from an affected father and the second copy of the mutant gene from their mother, who is most often a carrier (heterozygous) but who might be affected (homozygous). Males, on the other hand, have only one copy of the X chromosome, which they always receive from their mother. Therefore, males with an X chromosome-linked disease always receive the mutant copy of the gene from their mother. Moreover, because men don't have a second copy of the X chromosome to potentially "cancel out" the negative effects of X-linked mutations, they are far more likely than women to be affected by X chromosome-linked recessive diseases.
The blood-clotting disorder hemophilia A is one of several single-gene diseases that exhibit an X chromosome-linked recessive pattern of inheritance. Males who have a mutant copy of the factor VIII gene (F8) will always have hemophilia. In contrast, women are rarely affected by this disease, although they are most often carries of the mutated gene. Duchenne muscular dystrophy is another example of a single-gene disease that exhibits an X chromosome-linked recessive inheritance pattern. This condition is associated with mutations in the dystrophin gene (DMD).
X Chromosome–Linked Dominant Single-Gene Diseases
Few dominantly inherited forms of human disease are X chromosome linked. Females with an X chromosome-linked dominant disease can inherit the mutant gene from either an affected mother or an affected father, whereas males always inherit such diseases from an affected mother.
Examples of X chromosome-linked dominant diseases are rare, but several do exist. For instance, dominant mutations in the phosphate-regulating endopeptidase gene (PHEX), which resides on the X chromosome, are associated with X-linked dominant hypophosphatemic rickets. Similarly, Rett syndrome, a neurodevelopmental disease, is associated with dominant mutations in the methyl-CpG-binding protein 2 gene (MECP2). Rett syndrome almost exclusively affects females, because male embryos with a dominant mutation in the MECP2 gene rarely survive.
Y Chromosome–Linked Single-Gene Disease
Like X-linked dominant diseases, Y chromosome-linked diseases are also extremely rare. Because only males have a Y chromosome and they always receive their Y chromosome from their father, Y-linked single-gene diseases are always passed on from affected fathers to their sons. It makes no difference whether the Y chromosome-linked mutation is dominant or recessive, because only one copy of the mutated gene is ever present; thus, the disease-associated phenotype always shows.
One example of a Y-linked disorder is nonobstructive spermatogenic failure, a condition that leads to infertility problems in males. This disorder is associated with mutations in the ubiquitin-specific protease 9Y gene (USP9Y) on the Y chromosome.
Using the Human Genome Sequence to Study Disease
With the complete sequence of the human genome in hand, scientists are now poised to match monogenic disease phenotypes to their corresponding genes. By analyzing complex pedigrees, geneticists can correlate changes in gene sequence with particular disease states. After all, once a disease-associated change in the DNA sequence of a gene is identified, it is much easier to determine how the structure of the corresponding gene product (protein) might be changed in a manner that alters its biological function. The nature of disease-associated changes in protein structure and function can in turn enhance our ability to design drugs that effectively and specifically target mutant proteins.
Recent estimates predict that the human genome includes 25,000 protein-encoding genes. Although 1,822 of the protein-encoding genes in humans are estimated to be associated with monogenic disease, the identities of more than 1,500 of these genes remain unknown, largely because many of these single-gene diseases are rare and occur in small numbers of families (Antonarakis & Beckmann, 2006). Referred to as "orphan" diseases, these relatively uncommon disorders receive much less research funding than more common diseases, which are often considered a better investment by funding agencies and biopharmaceutical companies. However, many of the common diseases exhibit a more complex inheritance pattern and are associated with mutations in multiple genes (in other words, these conditions are polygenic). As a result, research efforts have begun to shift from a focus on monogenic disease to a focus on polygenic disease, which can involve complex interactions between genes and the environment that are not easily interpreted.
From Monogenic to Oligogenic Disease: Modifier Genes
In recent years, a number of diseases initially characterized as monogenic have been shown to be caused or modified by an additional gene or genes. These diseases have been categorized as "oligogenic" rather than "polygenic," because they involve only a relatively small number of genes. For example, cystic fibrosis is typically characterized as a single-gene disease associated with recessive mutations in the CFTR gene. However, more extensive studies of CFTR mutations in larger and more diverse populations have shown that mutations in additional genes could perhaps modulate the severity and type of disease-related phenotypes (Figure 1).
Collectively, studies of disease that start with the discovery of a single disease-associated gene can provide an invaluable opportunity to expand our knowledge of more complex oligogenic links through the discovery of additional causative or modifying genes.
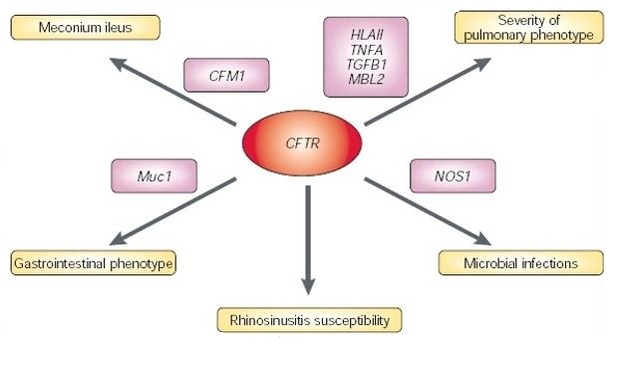
Figure 1: Complexity in monogenic diseases.
Mutations in CFTR almost always cause the CF phenotype. Owing to modification effects by other genetic factors, the presence and nature of mutations at the CFTR locus cannot predict what the phenotypic manifestation of the disease will be. Therefore, although CF is considered a Mendelian recessive disease, the phenotype in each patient depends on a discrete number of alleles at different loci. Meconium ileus describes the obstruction at birth of the small and/or large intestine (ileus) with the first fecal excretion (meconium). (NB. CFTR = cystic fibrosis transmembrane conductance regulator, CFM1 = cystic fibrosis modifier, HLA-II = MHC class II antigen, MBL2 = mannose-binding lectin (protein C) 2, NOS1 = nitric oxide synthase 1, TGFB1 = transforming growth factor-a1, and TNFA = tumour necrosis factor-a encoding gene.)
© 2002 Nature Publishing Group Badano, J. L. et al. Beyond Mendel: an evolving view of human genetic disease transmission. Nature Review Genetics 3, 780 (2002). All rights reserved. 
References and Recommended Reading
Antonarakis, S. E., & Beckmann, J. S. Mendelian disorders deserve more attention. Nature Reviews Genetics 7, 277–282 (2006) doi:10.1038/nrg1826 (link to article)
Badano, J. L., & Katsanis, N. Beyond Mendel: An evolving view of human genetic disease transmission. Nature Reviews Genetics 3, 779–789 (2002) doi:10.1038/nrg910 (link to article)
Guggino, W .B., & Stanton, B. A. New insights into cystic fibrosis: Molecular switches that regulate CFTR. Nature Reviews Molecular Cell Biology 7, 426–436 (2006) doi:10.1038/nrm1949 (link to article)
Jervis, G. A. Studies on phenylpyruvic oligophrenia: The position of the metabolic error. Journal of Biological Chemistry 169, 651–656 (1947)
Jervis, G. A. Phenylpyruvic oligophrenia deficiency of phenylalanine-oxidizing system. Proceedings of the Society for Experimental Biology and Medicine 82, 514–515 (1953)
Madeau, J. H. Modifier genes in mice and humans. Nature Reviews Genetics 2, 165–174 (2001) doi:10.1038/35056009 (link to article)
Paul, D. A double-edged sword. Nature 405, 515 (2000) doi:10.1038/35014676 (link to article)










