
Abstract
Coastal and demersal chondrichthyans, such as the small-spotted catshark, are expected to exhibit genetic differentiation in areas of complex geomorphology like the Mediterranean Basin because of their limited dispersal ability. To test this hypothesis, we used a fragment of the mitochondrial cytochrome c oxidase subunit I gene and 12 nuclear microsatellite loci in order to investigate the genetic structure and historical demography of this species, and to identify potential barriers to gene flow. Samples were collected from the Balearic Islands, the Algerian Basin, the Ionian Sea, the Corinthian Gulf and various locations across the Aegean Sea. Additional sequences from the Atlantic and the Levantine Basin retrieved from GenBank were included in the mitochondrial DNA analysis. Both mitochondrial and nuclear microsatellite DNA data revealed a strong genetic subdivision, mainly between the western and eastern Mediterranean, whereas the Levantine Basin shared haplotypes with both areas. The geographic isolation of the Mediterranean basins seems to enforce the population genetic differentiation of the species, with the deep sea acting as a strong barrier to its dispersal. Contrasting historical demographic patterns were also observed in different parts of the species' distribution, most notably a population growth trend in the western Mediterranean/Atlantic area and a slight decreasing one in the Aegean Sea. The different effects of the Pleistocene glacial periods on the habitat availability may explain the contrasting demographic patterns observed. The current findings suggest that the small-spotted catshark exhibits several genetic stocks in the Mediterranean, although further study is needed.
Similar content being viewed by others
Introduction
Sharks as top predators have an important ecological role in the marine environment, comprising at the same time one of the world’s largest fisheries resources (FAO, 2014). However, the growth of fisheries at global scale has led several species to come under threat, mainly because of their inherent characteristics such as slow growth, late maturity and low fecundity (Worm et al., 2013), which limit their capacity to recover from population declines (for example, Cailliet et al., 2005).
The inadequate scientific information on chondrichthyan population structure is often one of the reasons for failing to introduce suitable measures for their conservation (Cavanagh and Gibson, 2007). However, during the last two decades, significant progress has been made in our understanding of the historical and contemporary processes driving chondrichthyan population genetic patterns (Dudgeon et al., 2012). Recent studies of sharks and other chondrichthyans have shown that the level of intraspecific genetic differentiation appears to be highly correlated with species' dispersal ability, which is dependent on their maximum body size and habitat preference (coastal, pelagic or benthic) (Verissimo et al., 2010). Sharks, like all elasmobranchs, do not have pelagic eggs and larvae, and consequently their dispersal ability and potential to overcome barriers depend on actively swimming adults, being usually higher in large, pelagic and oceanic species (Musick et al., 2004). For example, large, oceanodromous species, such as the shortfin mako, Isurus oxyrinchus (Schrey and Heist, 2003) and the basking shark, Cetorhinus maximus (Hoelzel et al., 2006), exhibited genetic homogeneity across ocean basins. On the other hand, strong genetic structure was observed in less vagile, demersal species, such as the nurse shark, Ginglymostoma cirratum (Karl et al., 2012) and the whitetip reef shark, Triaenodon obesus (Whitney et al., 2012). Nonetheless, strong genetic structure has been reported even in highly vagile species, being attributed either to site fidelity, as for the sandbar shark, Carcharhinus plumbeus (Portnoy et al., 2010) or to environmental barriers, as for the spiny dogfish Squalus acanthias (Verissimo et al., 2010).
In the Mediterranean Sea, the dropping of the sea level during the glacial periods of the Pleistocene affected the hydraulic connectivity between basins by reducing water flow regimes among the western and eastern Mediterranean and the Atlantic. This phenomenon, together with changes in abiotic (for example, temperature and salinity) and biotic factors (for example, productivity), led many marine taxa to periods of extinction and recolonization, but also to genetic differentiation, which may have been retained because of past or present-day hydrographic barriers to gene flow (Patarnello et al., 2007; Pérez-Losada et al., 2007). Within the Mediterranean, different barriers to gene flow have been identified, such as the Almeria-Oran front formed east of the Strait of Gibraltar by the convergent Atlantic and Mediterranean water masses (Galarza et al., 2009), the Siculo-Tunisian Strait, which divides the eastern and western Mediterranean basins, and the hydrographic isolation of the Aegean, Ionian and Adriatic Seas (for example, Pérez-Losada et al., 2007 and references therein).
The small-spotted catshark, Scyliorhinus canicula (Linnaeus, 1758), is one of the smallest catshark species and the most abundant in the European inshore waters (Ellis and Shackley, 1997). It is distributed on the continental shelf and uppermost slope waters throughout the Mediterranean Sea, as well as in the northeast Atlantic Ocean, from Norway and the British Isles in the north, to Senegal and possibly along the Ivory Coast to the south (Compagno, 1984). It is characterized as a bottom dwelling species, occurring primarily over sandy, gravel or muddy bottoms from a few metres down to 500 m in depth (Compagno, 1984). Through internal fertilization and oviparity, it produces egg cases throughout the year (Kousteni et al., 2010), which are anchored to macroalgae and other solid structures (Wheeler, 1978). As an opportunistic scavenger, it interacts greatly with the substratum in order to feed on a wide range of benthic fauna (Mnasri et al., 2012). According to a tagging study, adults show high degree of site fidelity, whereby 70% of the recaptured individuals did not move >24 km and the maximum distance recorded was 256 km (Rodríguez-Cabello et al., 2004).
Although the species is listed as Least Concern (IUCN, 2014), it was considered overexploited in a recent stock assessment (Cardinale and Osio, 2013), whereas in some areas localized depletion of its populations has already been reported (Gibson et al., 2006). In general, it is a moderately important commercial species, which is primarily caught as bycatch in various demersal fisheries and retained for human consumption, oil extraction or fish bait (Compagno, 1984). It is also commonly displayed in public aquaria because of its relatively easy maintenance in captivity, while several thousand egg cases are produced annually to meet the needs of the international scientific community, which uses this species as a genomic model (Coolen et al., 2009).
Coastal, demersal and small-sized chondrichthyans with low dispersal potential, like the small-spotted catshark, are expected to exhibit genetic differentiation and low connectivity, especially in areas of complex geomorphology such as the Mediterranean Sea. However, a study of the thornback ray, Raja clavata, showed that the Mediterranean populations were undifferentiated, sharing only a single ancestral mitochondrial haplotype, and the respective analyses suggested a strong bottleneck and a subsequent isolation from the Atlantic (Chevolot et al., 2006). Griffiths et al. (2011a) did not find any shared haplotypes between Atlantic and Mediterranean samples of the longnose skate, Dipturus oxyrinchus, suggesting the genetic isolation of the Mediterranean from the other stocks. On the other hand, first indications for the genetic differentiation of the small-spotted catshark within the Mediterranean were recently found based on mitochondrial DNA (mtDNA) analysis (Barbieri et al., 2014).
Previous attempts to identify the small-spotted catsharks’ stocks in the Atlantic had relied on the spatial variation in natural marks, including parasites (Moore, 2001) and mercury concentrations (Leah et al., 1991), as well as on tagging studies (Rodríguez-Cabello et al., 2004), all of them suggesting localized populations. Moreover, the latitudinal variation on the species’ life history traits, namely, the increase of body size (for example, Leloup and Olivereau, 1951) and size at maturity (for example, Kousteni et al., 2010) from the Mediterranean to the Atlantic, suggests the presence of distinct stocks in these areas.
In this study, the population genetic structure and the historical demography of the small-spotted catshark were assessed in different spatial scales (within and between basins) by two types of genetic markers with different modes of inheritance: the mtDNA cytochrome c oxidase subunit I (COI) gene and 12 polymorphic nuclear microsatellite loci. Based on these data, we tested the hypothesis that small-spotted catsharks form several genetic stocks in the Mediterranean because of their life history traits in combination with the complex geomorphology of the region. We further tried to identify current and historical barriers to gene flow as well as explore the Pleistocene’s effects on the historical demography of the species. Finally, we discussed the implications of our findings for the management and conservation of this species.
Materials and methods
Sample collection and DNA extraction
Small-spotted catsharks were caught by commercial bottom trawlers and bottom longliners in the Mediterranean Sea between December 2005 and June 2012 at depths between 66 and 512 m. Sampling was carried out across the Mediterranean Sea, from the western to the eastern basins (Figure 1, Table 1). It was denser in the east where sample collections were obtained from different sub-basins within the Aegean. In this way, the population genetic structure was assessed on either side of the potential barriers to gene flow and at different spatial scales.

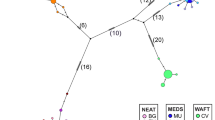
Map indicating the sampling sites of Scyliorhinus canicula collections included in the mtDNA analysis along with the haplotype median-joining network for the COI fragment. Each number represents a haplotype. Small black circles represent missing, probably unsampled or extinct haplotypes. Aegean (AEG) includes the sample collections from CRE, SAR, CYC, nAEG and nEVO. The sample collection codes are explained in Table 1.
Individual fin clips were preserved in 95% ethanol at room temperature. Total genomic DNA was extracted from approximately 25 mg of fin tissue following the salting-out protocol by Miller et al. (1988). The geographic origin and the number of specimens used in mtDNA and nuclear microsatellite data analyses are shown in Table 1. Thirty-nine additional sequences were included in the mtDNA analysis (Table 1): 17 from the Atlantic (ATL; accession numbers: JQ774728–JQ774732, JQ774911–JQ774914, JQ775119–JQ775126) by Costa et al. (2012) and 22 from the Levantine Basin (LEV; accession numbers: JN641222–JN641233, JN641234–JN641243) by Moftah et al. (2011).
PCR amplification and sequencing of mtDNA COI
A 738-bp fragment of the mtDNA COI gene was amplified for each of the 392 specimens by PCR using the M13-tailed primers FishF2_t1 and FishR2_t1 (Ward et al., 2005; Ivanova et al., 2007). The tailed primers were used to generate longer sequencing reads. PCR reactions were performed in 10 μl total volume containing: 1 μl DNA template, 5 μl trehalose (10%), 1.9 μl ultra-pure water, 1 μl 10x PCR buffer, 0.5 μl of MgCl2 (50 mM), 0.2 μl dNTPs (10 mM), 0.15 μl of each primer (10 μM) and 0.5 U Taq polymerase. Amplifications were performed in a T100 thermal cycler (Bio-Rad, Hercules, CA, USA) with an initial denaturation of 2 min at 95 °C, followed by 35 cycles of 30 s at 94 °C, 45 s at 51 °C and 45 s at 72 °C, and a final extension step for 10 min at 72 °C.
PCR products (1 μl) were visualized on agarose gels and the remaining volume was purified by ethanol precipitation. The forward and—in cases where the full sequence was not obtained—the reverse strands of the amplicons were sequenced using the ABI Big Dye Terminator Cycle Sequencing Kit (Applied Biosystems Inc., Foster City, CA, USA) and run on an ABI 3730xl DNA sequencer (Applied Biosystems Inc., Carlsbad, CA, USA). Each sequencing reaction mixture (total volume 6 μl) consisted of 0.3 μl Big Dye, 0.096 μl forward primer (10 μM), 1.05 μl 5x buffer and 10–20 ng of DNA template, and the remaining volume of the reaction was completed with ultra-pure water. Cycle sequencing reaction was performed with an initial denaturation of 3 min at 96 °C, followed by 40 cycles of 20 s at 96 °C, 15 s at 55 °C and 4 min at 60 °C, and final incubation at 4 °C.
The mtDNA COI sequences were imported in BioEdit v7.2 (Ibis Therapeutics, Carlsbad, CA, USA) and checked by eye for quality and accuracy in nucleotide base assignment. All the sequences were aligned with the CLUSTAL W algorithm (Higgins et al., 1994), using the software MEGA v5.1 (Kumar et al., 2008), and were trimmed to 590 bp for further analysis. MEGA was used to identify singleton mutations and parsimony informative sites. The 392 sequences obtained in this study were deposited in GenBank (accession numbers: KM819715–KM820106) and, together with 39 previously published sequences (Moftah et al., 2011; Costa et al., 2012), represented 11 sample collections according to their geographic origin (Table 1).
PCR amplification and genotyping of microsatellite loci
A total of 834 individuals were genotyped at 12 microsatellite loci (Scan02, Scan03, Scan04, Scan06, Scan09, Scan10, Scan11, Scan12, Scan13, Scan14, Scan15 and Scan16) described in Griffiths et al. (2011b). The loci were chosen after testing for consistent amplification. The forward primers were fluorescently labelled with FAM, HEX or ROX. All 12 loci were amplified together in a single multiplex PCR reaction using the QIAGEN Multiplex PCR Kit (Qiagen, Chatsworth, CA, USA) and protocol, and electrophoresed on an ABI 3730xl along with the GeneScan 500 LIZ size standard (Applied Biosystems Inc., Foster City, CA, USA). PCR reactions were repeated for several (16.07%) individuals in order to fill in missing data and to confirm genotypes.
Genotypes were scored manually with the software STRand v2.4.59 (Veterinary Genetics Laboratory, University of California, Davis, CA, USA). The software MICROCHECKER v2.2.3 (Van Oosterhout et al., 2004) was used to check for null alleles, large allele dropout and stutter peaks. All 834 individual genotypes were finally grouped into 10 sample collections according to their geographic origin (Table 1).
Genetic diversity
The genetic variability of mtDNA COI sequences (number of singleton and parsimony informative sites) and the genetic p-distance among haplotypes were estimated in MEGA. Molecular diversity indices for the mtDNA COI fragment, including the number of haplotypes (Hap), the number of polymorphic sites (P), haplotype diversity (h), nucleotide diversity (π) and the mean number of nucleotide differences between haplotypes (k), were calculated for each sample collection using the software ARLEQUIN v3.5.1.2 (Excoffier et al., 2005).
For the nuclear microsatellite data, deviations from Hardy–Weinberg equilibrium and tests for linkage disequilibrium were performed in GENEPOP v4.2 (Raymond and Rousset, 1995) with Markov chain lengths obtained with the parameters: dememorization number=10 000, number of batches=100, number of iterations per batch=5000. Indices of genetic diversity including the number of alleles per locus (A), observed heterozygosity (HO) and expected heterozygosity (HE) were calculated for each locus and sampling location using the software ARLEQUIN. Allelic richness (AR) was calculated by the software FSTAT v2.9.3.2 (Goudet, 2001). The inbreeding coefficient (FIS) and its significance value for each sample collection were estimated using the software GENEPOP. Sequential Bonferroni correction was used to adjust the significance levels (Rice, 1989).
Population differentiation and structure
The relationships of the mtDNA haplotypes were depicted with a median-joining network (Bandelt et al., 1999) that was constructed in the software NETWORK v.4.6.1.1 (Fluxus Technology Ltd, Suffolk, UK).
The level of among-population genetic differentiation was estimated in the software ARLEQUIN by calculating the pairwise ΦST values for the mtDNA, based on the mean number of pairwise differences among sequences and the pairwise FST values for the nuclear microsatellite data based on allelic frequencies. Statistical significance was assessed using 1000 permutations of the data and sequential Bonferroni correction was used to adjust the significance levels (Rice, 1989).
The hierarchical structure of populations was assessed for each type of marker by analysis of molecular variance implemented in ARLEQUIN. For mtDNA, a Kimura two-parameter genetic distance matrix between haplotypes was used, whereas for nuclear microsatellite data, analysis of molecular variance was performed on a locus-by-locus basis and integrated over all the loci, based on allelic frequency data. Three different sample collection groupings were tested, considering their geographic origin and their genetic differentiation inferred by the ΦST and FST values: (a) one group, including all samples that accounted for one panmictic population, (b) two groups, the first including the western Mediterranean (plus the ATL for mtDNA) and the second including the eastern Mediterranean, and (c) three groups, the first including the western Mediterranean (plus the ATL for mtDNA), the second including the Ionian Sea (ION) and the Corinthian Gulf (COR) and the third including the remaining sampling sites from the eastern Mediterranean. For both markers, the statistical significance was estimated using 1000 permutations.
A neighbour-joining tree of the populations was constructed based on the microsatellite allele frequency data using the software POPTREE v2.0 (Takezaki et al., 2010). Nei’s DA genetic distance measure (Nei et al., 1983) was used with 1000 bootstrap replications.
The population structure was also assessed using a Bayesian model-based clustering approach implemented in STRUCTURE v.2.3 (Pritchard et al., 2000). This programme builds genetic clusters by minimizing linkage disequilibrium and deviations from Hardy–Weinberg equilibrium expectations. All the individuals were assigned to clusters without prior knowledge of their geographic origin using the admixture model with correlated allelic frequencies. Five independent runs with a number of potential genetic clusters (K) from one to five were carried out to verify that the estimates of K were consistent across runs. The burn-in length was set at 250 000 iterations followed by a run phase of one million iterations. The generated results were imported into the software STRUCTURE HARVESTER (Earl and von Holdt, 2012) to calculate the ad hoc ΔK statistic (Evanno et al., 2005). The K value, where ΔK had the highest value was identified as the most likely number of clusters. The results from STRUCTURE analysis were summarized by creating a histogram plot in MS EXCEL 2007, where each individual was represented by a single vertical line broken into K coloured segments with lengths proportional to each of the K inferred clusters (Pritchard et al., 2000).
Demographic history
Three commonly used methods were applied on the mtDNA data set to explore the demographic history of the different small-spotted catshark sample collections that seemed to constitute distinct evolutionary units.
First, deviations from selective neutrality and evidence for population expansion were tested with Fu’s Fs estimator (Fu, 1997) implemented in ARLEQUIN. Under demographic expansion, this index is expected to have significant negative values.
Second, evidence of population expansion was tested by mismatch distribution analysis implemented in ARLEQUIN. This method is based on the assumption that population growth or decline leaves distinctive signatures in the DNA sequences compared with a constant population size (Rogers and Harpending, 1992). A mismatch distribution was constructed by comparing the observed frequency distribution of pairwise nucleotide differences between haplotypes to that expected under a sudden population expansion model (Schneider and Excoffier, 1999). The mismatch distribution is expected to be multimodal in stable populations and unimodal in recently expanding populations. The Harpending’s raggedness index (r), quantifying the smoothness of the observed mismatch distributions, was computed to determine the goodness of fit to a unimodal distribution (Harpending, 1994). The statistical significance was tested using a bootstrap approach (1000 permutations). A significant raggedness index value (Pr<0.05) was taken as evidence for rejecting the sudden population expansion model (Schneider and Excoffier, 1999). The time at which population expansion began (t) was calculated from the equation τ=2μt, where τ is the mode of the mismatch distribution and μ is the mutation rate for the entire sequence. For DNA sequence data covering k nucleotides, the mutation rate (μ) equals to: μ=ku, where u is the mutation rate per nucleotide (Rogers and Harpending, 1992). Given the lack of a species-specific mutation rate for the COI gene, the substitution rate (2.38 × 10−9 substitutions per site per year) that was reported for the COI gene in angel sharks (Stelbrink et al., 2010) was used.
Third, in order to reconstruct the past population dynamics of the small-spotted catshark, a Bayesian skyline plot model was computed in the software BEAST v1.7.5 (Drummond et al., 2012). A strict molecular clock was assumed under an HKY+I substitution model of molecular evolution determined by jMODELTEST v.2.1.4 (Darriba et al., 2012). Each subset was run twice for 100 million iterations of the Markov chain Monte Carlo chains, following a burn-in length of 10 million iterations and sampled every 1000 steps, starting from an UPGMA tree and applying default Bayesian priors. The Bayesian skyline reconstructions were conducted in the software TRACER v.1.5 (Rambaut and Drummond, 2007). The time to the most recent common ancestor (tMRCA) was inferred for each Bayesian skyline plot model.
Results
Genetic diversity
Out of 431 mtDNA sequences, 37 haplotypes were identified based on 32 polymorphic sites (11 singleton and 21 parsimony informative sites) (Supplementary Table S1). Ten of these haplotypes had been previously identified by Costa et al. (2012) in the ATL (hap1, 4, 5, 7, 8, 14) and by Moftah et al. (2011) in the LEV (hap1, 2, 4, 6, 12, 13). The genetic p-distance between haplotypes was low and ranged from 0.17 to 1.36%. The overall haplotype (h) and nucleotide (π) diversity were 0.808±0.012 and 0.0032±0.002, respectively. The COR showed the lowest haplotype (0.439±0.158) and nucleotide (0.0011±0.001) diversity, whereas the LEV showed the highest haplotype (0.9±0.036) and nucleotide (0.0043±0.0027) diversity. The mean number of nucleotide differences between haplotypes within populations (k) ranged from 0.64 (COR) to 2.52 (LEV) (Table 2).
The analysis of the 12 microsatellite loci showed no evidence for null alleles, short allele dominance or stuttering. Pairwise comparisons between loci revealed no linkage disequilibrium, whereas departures from Hardy–Weinberg equilibrium were detected only in locus Scan02 in one sample (ION) and in locus Scan03 in two samples (ION and Cyclades Islands (CYC)) (Supplementary Table S2). Genetic diversity (measured as observed heterozygosity) was similar among samples, ranging from 0.54 (Cretan Sea (CRE)) to 0.64 (Algerian Basin (ALG)), whereas allelic richness ranged from 4.62 (COR) to 5.63 (ALG) (Table 2).
Phylogeographic analysis
The haplotype network had a star-like shape and consisted of a central haplotype (hap1) that was observed in all sampling sites and was shared by 34% of the specimens (Figure 1). The rest of the haplotypes were found either in the western Mediterranean/Atlantic or in the eastern Mediterranean. The LEV was an exception, as it contained haplotypes from both the aforementioned areas. Apart from the central haplotype, there were four other abundant haplotypes (>5% frequency in the whole data set): two of them (hap2 and 3) found in the eastern Mediterranean, and the other two (hap4 and 5) in the western Mediterranean/Atlantic. Haplotypes 2 and 4 were also found in the LEV. In total, 12 haplotypes were found in more than one location and 25 were private (20 of which were singletons) (Supplementary Table S1).
Population differentiation and structure
For mtDNA, pairwise ΦST values ranged from 0 to 0.516 and were significant for all pairwise comparisons between the western Mediterranean/Atlantic and eastern Mediterranean sample collections, except between ATL and LEV (Table 3). Within the western Mediterranean/Atlantic and the eastern Mediterranean, pairwise ΦST values were not significant.
For nuclear microsatellite data, pairwise FST values ranged from 0 to 0.100 (Table 3). Pairwise FST estimates were significant among collections from the western (Balearic Islands (BAL), ALG) vs eastern Mediterranean (all other collections). All comparisons including either the ION or the COR were also significant, except ION vs COR, which was not significantly different. In the western Mediterranean, BAL and ALG were also significantly different. Within the Aegean Sea, the pairwise FST values ranged from 0 to 0.009 and were not significant, except between north Aegean Sea (nAEG) and Myrtoan Sea (MYR).
The results of the analysis of molecular variance analysis were generally concordant between mtDNA and microsatellites (Supplementary Table S3). The null hypothesis of global panmixia was rejected because of significant genetic heterogeneity among various sampling sites (ΦST=0.263, FST=0.040, P<0.001). The highest among group differentiation (ΦCT=0.409, P<0.01 for mtDNA and FCT=0.064, P<0.05 for microsatellites) was found when considering two main groups: the first including all samples from the western Mediterranean (plus ATL for mtDNA analysis) and the second including all samples from the eastern Mediterranean (plus LEV for mtDNA analysis).
The neighbour-joining tree of populations based on Nei’s DA genetic distance from allelic frequency data (Figure 2) indicated that the western Mediterranean samples formed one group, all the Aegean samples formed another one, whereas the ION and the COR had intermediate positions between the Mediterranean and the Aegean groups.

Neighbouring-joining tree based on Nei’s genetic distances DA generated from nuclear microsatellite allele frequencies of Scyliorhinus canicula sample collections with bootstrap values estimated from 1000 replications (a). Genetic clustering of Scyliorhinus canicula sample collections based on 12 microsatellite loci for the best K=2, generated from STRUCTURE analysis. The collections are separated by black vertical lines. Each individual is represented by a narrow vertical column broken into K=2 coloured segments with lengths proportional to each of the inferred K clusters (b). The sample collection codes are explained in Table 1.
The genetic subdivision between the western and eastern Mediterranean that was inferred from the previous analyses is also consistent with the number of clusters identified by STRUCTURE. Based on the microsatellite data, STRUCTURE revealed the highest likelihood at K=2 clusters (average log probability of data Ln[P(D|K)]=−25191.43±1.23 s.d.), indicating this as the best estimate of the true number of genetic clusters (Supplementary Figure S1). Both clusters were represented in all 10 populations, but with different proportions (Figure 2). Both the BAL and the ALG (western Mediterranean) exhibited high population homogeneity with the lowest proportion of ancestry in cluster 1 (3% and 12%, respectively), whereas the locations from the Aegean Sea had the highest proportion in this cluster (79–90%). The ION had almost equal proportions to both clusters (55% in cluster 1) and the COR had a higher proportion in cluster 1 (74%).
Demographic history
Fu’s neutrality test and the mismatch distribution analysis were performed for the whole mtDNA data set, as well as separately for the western Mediterranean/Atlantic, Ionian and Aegean collections, considering the patterns revealed by pairwise ΦST (Table 4). Based only on the mismatch distribution analysis, all the sample collections conformed to the model of sudden population expansion (Figure 3). However, the statistically significant negative Fu’s index, in combination with the nonsignificant raggedness index, indicated a sudden population expansion scenario only for the western Mediterranean/Atlantic sample collections.

Haplotype mismatch distributions based on the observed and expected pairwise nucleotide differences between haplotypes of Scyliorhinus canicula according to the sudden expansion model (a). Bayesian skyline plots derived from COI sequences (mtDNA) for the western Mediterranean/Atlantic, ION, Aegean Sea and all sampled populations. The x axis represents the time since the present in years and the y axis represents the estimated effective population size multiplied by the generation time. The thick solid line is the median estimate, and the dotted lines show the 95% HPD limits. The time to the most recent common ancestor is shown by a dashed vertical line (b).
According to the population expansion parameter (τ) and given the assumed mutation rate, the population expansion of the western Mediterranean/Atlantic sample was estimated at about 574 thousand years before the present (ky BP) (464–734 ky BP at 95% confidence interval). This estimate is broad, but suggests a scenario of Middle Pleistocene population expansion for the small-spotted catshark in the western Mediterranean (Table 4).
The Bayesian Skyline plots also suggested a sudden population growth of the western Mediterranean/Atlantic group, which was estimated to have started slightly later (at about 450 ky BP) than predicted by the mismatch distribution. However, it also supported a Middle Pleistocene population expansion event. The pattern for the Ionian and the Aegean collections was that of a stable population showing a slight increasing trend for the Ionian and a slight decreasing trend for the Aegean population (Figure 3). It seems that the sudden population expansion scenario is supported by all three demographic analyses only in the case of the western Mediterranean/Atlantic sample collections.
Discussion
Population structure and barriers to gene flow
Strong genetic structure was revealed for the small-spotted catshark, Scyliorhinus canicula, by both mitochondrial and nuclear microsatellite data analyses. This structure can be attributed to the low dispersal ability of the species because of its life history traits, especially the preference for coastal and benthic habitats, the lack of pelagic developmental stages (Compagno, 1984), and the high degree of site fidelity (Rodríguez-Cabello et al., 2004). Considering the species’ characteristics, we can assume that the deeper marine basins constitute a strong natural barrier to its dispersal. This notion is supported by the different levels of genetic differentiation observed between locations across similar spatial scales that are separated by different depths. There was very little differentiation within a distance of ~300 km in the Aegean Sea (max FST=0.009), which is mainly a shallow plateau that provides a relatively continuous habitat for the small-spotted catshark. In contrast, in the western Mediterranean there was significant divergence between the sample collections from the Balearic Islands and the Algerian Basin (FST=0.032), which are <300 km apart but are separated by the Balearic Abyssal Plain. The effect of deep marine basins as a barrier to dispersal has also been demonstrated for other demersal chondrichthyans, such as the nurse shark, between the Brazilian coast and the offshore islands (Karl et al., 2012), and the thornback ray, between the European continental shelf and the Azores (Chevolot et al., 2006).
The genetic subdivision of the small-spotted catshark sample collections was mainly observed between the western and eastern Mediterranean. It was especially pronounced in the phylogeographic structure of the mtDNA haplotypes, where a central ancestral haplotype was found in all samples, whereas the derived haplotypes were present either in the western Mediterranean/Atlantic or in the eastern Mediterranean. An exception to this pattern was the Levantine Basin, which shared haplotypes with both the aforementioned areas. This indicates that the Siculo-Tunisian Strait is not an impermeable barrier to gene flow for this species, although its narrowing during the Pleistocene glacial periods probably enforced the genetic differentiation between the western and eastern Mediterranean. The disconnected position of the shared haplotypes of the Levantine Basin in the phylogenetic network suggests a historical gene flow from the northeastern and western Mediterranean to this area. This probably occurred along the coast, as the deep sea seems to be a natural barrier to the species' dispersal.
Further genetic differentiation, but only at the nuclear level, was observed within the western (BAL vs ALG) and eastern (ION and COR vs all Aegean sample collections) Mediterranean. The ION and COR samples had an intermediate position between the western Mediterranean and the Aegean in the nuclear microsatellite analysis, but not in the mtDNA analysis, as they shared haplotypes (apart from the central) only with the Aegean sample. The genetic isolation of both the Aegean and Ionian Seas from the western Mediterranean probably reflects their geographic isolation through the deep Hellenic Trench (>4000 m), lying along the western and southwestern Hellenic coast (Stergiou et al., 1997). In several marine species, the major genetic break or limitation to dispersal between the western and eastern Mediterranean was also related to the isolation of the Ionian and Aegean Seas, and not to the Siculo-Tunisian Strait (Borrero-Pérez et al., 2011 and references therein).
On the other hand, limited genetic connectivity between the Ionian and Aegean Seas was revealed with occasional dispersal events from one area to the other. For instance, haplotype 3 was found almost exclusively in the Aegean and in low frequency in the Ionian, indicating a dispersal event from the former to the latter. At the same time, haplotype 2 was found in high frequency in both areas, and it was connected to the ancestral haplotype through haplotype 16, which was present only in the Ionian Sea. The latter may indicate dispersal from the Ionian to the Aegean. However, this could also be explained by genetic drift because of habitat fragmentation and population decline.
Finally, the Corinthian Gulf, which is today a deep inlet (935 m) of the Ionian Sea separating the Peloponnese from the Greek mainland, was a lake during the last glacial maximum, when the sea level was about 120 m lower than present (Sakellariou et al., 2010). Based on radiochronology data, it was shown that sea water entered the former lake of the Corinthian Gulf through shallow straits about 13–14 ky BP (Sakellariou et al., 2010). Hence, the small-spotted catshark was probably established there during the Holocene from the Ionian Sea, when these areas were connected through the neighbouring Patraikos Gulf. To the east, the Corinthian Gulf was delimited from the Aegean Sea by the homonymous Isthmus until 1893, when a canal was constructed. The fact that one of the three haplotypes that were found in the COR was present in the Aegean (hap10), but not in the ION, may indicate very recent gene flow from the Aegean to the Corinthian Gulf. This assumption is further supported by the higher partition of the COR collection to cluster 1 (dominated by the Aegean sample collections) compared to the ION collection (Figure 2), in STRUCTURE analysis. Nevertheless, the sample size for the COR is relatively small for a safe conclusion to be drawn.
The observed genetic structure in this study is not congruent with previous findings by Barbieri et al. (2014) using mtDNA sequences alone. In that study, the western (Catalan, Ligurian and Tyrrhenian Seas) and eastern (Strait of Sicily and Levantine Basin) Mediterranean were not significantly divergent. However, genetic differentiation was detected between the Atlantic and the western Mediterranean, as well as between the Adriatic and the eastern Mediterranean. These differences may be attributed mainly to the different sampling scheme that was used.
Comparison with other chondrichthyan species of similar distribution and life history
The population genetic pattern found for the small-spotted catshark is not congruent with findings in the thornback ray (Chevolot et al., 2006) and the longnose skate (Griffiths et al., 2011a), although they have a similar geographic distribution and life history traits (Compagno, 1984). For the thornback ray, which also exhibits high degree of site fidelity (Hunter et al., 2005), a single mitochondrial haplotype was identified in the Mediterranean, attributed to a relatively recent expansion following a bottleneck (Chevolot et al., 2006). A similar pattern may have occurred for the small-spotted catshark much earlier in the past, as is suggested by the presence of a widespread, ancestral mitochondrial haplotype in high frequency in all the small-spotted catshark collections (which is consistent with a strong historical bottleneck followed by expansion). This may have happened either during a glacial period of the Pleistocene with the formation of a refugial population that at a later interglacial period expanded throughout the Mediterranean, or during a colonization event of the Mediterranean from the Atlantic. After the expansion, subsequent barriers to gene flow produced the current genetic differentiation. In conclusion, the different genetic patterns observed between the two species could reflect similar phylogeographic events that occurred at different periods of historical time.
In the small-spotted catshark, the lack of significant genetic differentiation between the Atlantic and the western Mediterranean at the mtDNA level contrasts the strong genetic differentiation that was detected in the longnose skate (Griffiths et al., 2011a). Griffiths et al. (2011a) found no shared haplotypes between the Atlantic and the Mediterranean collections, possibly because the Atlantic collections (Rockall Bank and off Norway) were geographically very distant from the Mediterranean ones (Mallorca and Menorca). However, the genetic differentiation of both species between the Atlantic and the Mediterranean remains to be clarified with more markers (for example, nuclear) and a more appropriate sampling scheme.
Nevertheless, a comparative analysis of several bony fishes in the Mediterranean failed to show any particular association between biological traits and population genetic patterns, even in closely related species with similar life history characteristics and dispersal abilities (Patarnello et al., 2007). In any case, the stochastic nature of the historical events could explain these discrepancies.
Demographic history
Apart from the strong population structure, contrasting patterns of demographic history of the small-spotted catshark were detected in different areas. In the western Mediterranean/Atlantic, a pattern of sudden population growth was detected that started in the Middle Pleistocene. This finding is in accordance with similar estimations for several chondrichthyan species (Chevolot et al., 2006 and references therein). On the other hand, the species exhibited a pattern of slight declining population size in the Aegean Sea and a slight increasing trend in the Ionian Sea.
These spatial differences in the demographic history of the small-spotted catshark could be attributed to the respective differences in favourable conditions and/or habitat availability during the Pleistocene glacial–interglacial cycles that caused dramatic shifts of the European coastline.
The Aegean Sea is a semi-closed basin fragmented by thousands of islands, delimited to the south by very deep marine basins and connected to the neighbouring areas by narrow strips of continental shelf. During the glacial periods of the Middle Pleistocene (425–250 ky BP), almost 50–60% of this area became land, with extensive drainage systems, deltaic plains and large lakes in the central and north parts (Lykousis, 2009). A similar situation also occurred during the last glacial period (Perissoratis and Conispoliatis, 2003). Consequently, the available habitat of the small-spotted catshark was severely restricted, with negative effects on its populations. Moreover, the post-glacial recovery of its populations was probably not so intense—because of the restricted area of the Aegean Sea—as to create a pattern of demographic expansion. These events probably explain the general historical demographic pattern of a slightly declining population in the Aegean Sea, which is also reflected in the low haplotype diversity detected in the respective sample collections (only six haplotypes were found in a total of 233 individuals sequenced).
The Pleistocene glacial–interglacial periods, probably also affected the Ionian Sea, which is an elongated embayment of the Mediterranean Sea and covers a narrow continental shelf along the west Hellenic coast (Perissoratis and Conispoliatis, 2003). However, it communicates with the shallow region of south Adriatic and south Italy, providing an area for population expansion. It is also less fragmented by islands compared to the Aegean. Thus, the effects of glaciations on the habitat and the population sizes of the small-spotted catshark may have been less severe in the Ionian Sea.
In the western Mediterranean, the historical population growth of the small-spotted catshark that contributed to its higher haplotype diversity may be explained by the less fragmented seascape, which was consequently less affected by the glacial periods and/or by the communication with the Atlantic Ocean. The latter may have served either as a new area for colonization during the interglacial periods or as a refuge during the glacial ones. Actually, the high haplotype diversity in the Atlantic sample may be consistent with the existence of a Pleistocene refuge along the Iberian Peninsula as it was also found for the thornback ray (Chevolot et al., 2006), although more samples are needed to test this hypothesis.
Conservation and management implications
This study indicates that the small-spotted catshark exhibits distinct genetic stocks within the Mediterranean, although more extended sampling is needed for their precise identification and delineation. According to our data, four distinct genetic stocks can be distinguished: off the Balearic Islands, off Algeria, as well as in the Ionian and Aegean Seas, which could be considered as separate management units. The finding that deep marine basins are a strong natural break in the species’ genetic connectivity should also be taken into account for future conservation and management plans, as populations in semi-isolated areas, such as the Aegean Sea, may be more vulnerable to over-exploitation and more difficult to recover after collapse.
Data archiving
Sequence data have been submitted to GenBank: accession numbers KM819715–KM820106. Genotype data have been submitted to Dryad: http://dx.doi.org/10.5061/dryad.nf61r
Accession codes
Accessions
GenBank/EMBL/DDBJ
References
Bandelt HJ, Forster P, Rohl A . (1999). Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16: 37–48.
Barbieri M, Maltagliati F, Roldánb MI, Castelli A . (2014). Molecular contribution to stock identification in the small-spotted catshark, Scyliorhinus canicula (Chondrichthyes, Scyliorhinidae). Fish Res 154: 11–16.
Borrero-Pérez GH, González-Wangüemert M, Marcos C, Pérez-Ruzafa A . (2011). Phylogeography of the Atlanto-Mediterranean sea cucumber Holothuria (Holothuria) mammata: the combined effects of historical processes and current oceanographical pattern. Mol Ecol 20: 1964–1975.
Cailliet GM, Musick JA, Simpfendorfer CA, Stevens JD . (2005). Ecology and life history characteristics of chondrichthyan fish. In: Fowler SL, Cavanagh RD, Camhi M, Burgess GH, Fordham SV, Simpfendorfer CA, Musick JA (eds). Sharks, Rays and Chimaeras: The Status of the Chondrichthyan Fishes. IUCN, Gland: Switzerland and Cambridge, UK.
Cardinale M, Osio GC . (2013) State of Fish Stocks and Fisheries in European Waters: Status of Mediterranean and Black Sea Resources in European Waters in 2013. European Comission Joint Research Center: Ispra.
Cavanagh RD, Gibson C . (2007) Overview of the Conservation Status of Cartilaginous fFshes (Chondrichthyans) in the Mediterranean Sea. Gland: Switzerland and Malaga, Spain. VI + pp 42.
Chevolot M, Hoarau G, Rijnsdorp AD, Stam WT, Olsen JL . (2006). Phylogeography and population structure of thornback rays (Raja clavata L., Rajidae). Mol Ecol 15: 3693–3705.
Compagno LJV . (1984). FAO species catalogue. Vol. 4. Sharks of the world. An annotated and illustrated catalogue of shark species known to date. Part 2. Carchariniformes. FAO Fisheries Synopsis 4: 251–265.
Coolen M, Menuet A, Chassoux D, Compagnucci C, Henry S, Lévèque L et al. (2009). The dogfish Scyliorhinus canicula, a reference in jawed vertebrates. In: Behriger RR, Johnson AD, Krumlauf RE (eds). Emerging Model Organisms. A Laboratory Manual Vol 1, Cold Spring Harbor: New York. pp 431–446.
Costa FO, Landi M, Martins R, Costa MH, Costa ME, Carneiro M et al. (2012). A ranking system for reference libraries of DNA barcodes: Application to marine fish species from Portugal. PLoS ONE 7: e35858.
Darriba D, Taboada GL, Doallo R, Posada D . (2012). jModelTest 2: more models, new heuristics and parallel computing. Nat Methods 9: 772.
Drummond AJ, Suchard MA, Xie D, Rambaut A . (2012). Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol 29: 1969–1973.
Dudgeon CL, Ovenden JR, Blower DC, Broderick D, Giles JL, Holmes BJ et al. (2012). A review of the application of molecular genetics for fisheries management and conservation of sharks and rays. J Fish Biol 80: 1789–1843.
Earl DA, von Holdt BM . (2012). STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Res 4: 359–361.
Ellis JR, Shackley SE . (1997). The reproductive biology of Scyliorhinus canicula in the Bristol Channel, UK. J Fish Biol 51: 361–372.
Evanno G, Regnaut S, Goudet J . (2005). Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14: 2611–2620.
Excoffier L, Laval G, Schneider S . (2005). Arlequin ver. 3.0: an integrated software package for population genetics data analysis. Evol Bioinform Online 1: 47–50.
FAO. (2014). Fisheries and Aquaculture Information and Statistics Service. Downloaded on 15 August 2014.
Fu YX . (1997). Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147: 915–925.
Galarza JA, Carreras-Carbonell J, Macpherson E, Pascual M, Roques S, Turner GF, Rico C . (2009). The influence of oceanographic fronts and early-life-history traits on connectivity among littoral fish species. Proc Natl Acad Sci USA 106: 1473–1478.
Gibson C, Valenti SV, Fowler SL, Fordham SV . (2006). The Conservation Status of Northeast Atlantic Chondrichthyans; Report of the IUCN Shark Specialist Group Northeast Atlantic Regional Red List Workshop. VIII + pp. 76.
Goudet J . (2001). FSTAT: a program to estimate and test gene diversities and fixation indices (version 2.9.3.2). Available at http://www.unil.ch/izea/softwares/fstat.html Updated from Goudet (1995).
Griffiths A, Sims D, Johnson A, Lynghammar A, McHugh M, Bakken T, Genner M . (2011a). Levels of connectivity between longnose skate (Dipturus oxyrinchus in the Mediterranean Sea and the north-eastern Atlantic Ocean. Conserv Genet 12: 577–582.
Griffiths AM, Casane D, McHugh M, Wearmouth VJ, Sims DW, Genner MJ . (2011b). Characterization of polymorphic microsatellite loci in the small-spotted catshark (Scyliorhinus canicula L.). Conserv Genet Res 3: 705–709.
Harpending RC . (1994). Signature of ancient population growth in a low-resolution mitochondrial DNA mismatch distribution. Hum Biol 66: 591–600.
Higgins D, Thompson J, Gibson T, Thompson JD, Higgins DG, Gibson TJ . (1994). CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22: 4673–4680.
Hoelzel AR, Shivji MS, Magnussen J, Francis MP . (2006). Low worldwide genetic diversity in the basking shark (Cetorhinus maximus. Biol Letters 2: 639–642.
Hunter E, Buckley AA, Stewart C, Metcalfe JD . (2005). Migratory behaviour of the thornback ray, Raja clavata, in the southern North Sea. J Mar Biol Ass UK 85: 1095–1105.
IUCN. (2014). IUCN Red List of Threatened Species. Version 2014.2 <www.iucnredlist.org> Downloaded on 31 July 2014.
Ivanova NV, Zemlak TS, Hanner RH, Hebert PDN . (2007). Universal primer cocktails for fish DNA barcoding. Mol Ecol Notes 7: 544–548.
Karl SA, Castro ALF, Garla RC . (2012). Population genetics of the nurse shark (Ginglymostoma cirratum in the western Atlantic. Mar Biol 159: 489–498.
Kousteni V, Kontopoulou M, Megalofonou P . (2010). Sexual maturity and fecundity of Scylliorhinus canicula (Linnaeus, 1758) in the Aegean Sea. Mar Biol Res 6: 390–398.
Kumar S, Nei M, Dudley J, Tamura K . (2008). MEGA: A biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief Bioinform 9: 299–306.
Leah RT, Evans SJ, Johnson MS, Collings S . (1991). Spatial patterns in accumulation by fish from the NE Irish Sea. Mar Pollut Bull 22: 172–175.
Leloup J, Olivereau M . (1951). Données biométriques comparatives sur la rousette (Scyllium canicula L.) de la Manche et de la Méditerranée. Vie Milieu 2: 182–209.
Lykousis V . (2009). Sea-level changes and shelf break prograding sequences during the last 400 ka in the Aegean margins: Subsidence rates and palaeogeographic implications. Cont Shelf Res 29: 2037–2044.
Miller SA, Dykes DD, Polesky HF . (1988). A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acid Res 16: 1215.
Mnasri N, El Kamel O, Boumaiza M, Reynaud C, Capapé C . (2012). Food and feeding habits of the small-spotted catshark, Scyliorhinus canicula (Chondrichthyes: Scyliorhinidae) from the northern coast of Tunisia (central Mediterranean). Cah Biol Mar 53: 139–150.
Moftah M, Abdel Aziz SH, Elramah S, Favereaux A . (2011). Classification of sharks in the Egyptian Mediterranean waters using morphological and DNA barcoding approaches. PLoS ONE 6: e27001.
Moore ABM . (2001). Metazoan parasites of the lesser-potted dogfish Scyliorhinus canicula and their potential as stock discrimination tools. J Mar Biol Ass UK 81: 1009–1013.
Musick JA, Harbin MM, Compagno LJV . (2004). Historical zoogeography of the Selachii. In: Carrier JC, Musick JA, Heithaus MR (eds) Biology of Sharks and Their Relatives. CRC Press: Boca Raton, Florida. pp 33–78.
Nei M, Tajima F, Tateno Y . (1983). Accuracy of estimated phylogenetic trees from molecular data. II. Gene frequency data. J Mol Evol 19: 153–170.
Patarnello T, Volckaert FAM, Castilho R . (2007). Pillars of Hercules: is the Atlantic-Mediterranean transition a phylogeographical break? Mol Ecol 16: 4426–4444.
Perissoratis C, Conispoliatis N . (2003). The impacts of sea-level changes during latest Pleistocene and Holocene times on the morphology of the Ionian and Aegean seas (SE Alpine Europe). Mar Geol 196: 146–156.
Portnoy DS, McDowell JR, Heist EJ, Musick JA, Graves JE . (2010). World phylogeography and male-mediated gene flow in the sandbar shark, Carcharhinus plumbeus. Mol Ecol 19: 1994–2010.
Pritchard JK, Stephens M, Donnelly P . (2000). Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
Pérez-Losada M, Nolte MJ, Crandall KA, Shaw PW . (2007). Testing hypotheses of population structuring in the Northeast Atlantic ocean and Mediterranean sea using the common cuttlefish Sepia officinalis. Mol Ecol 16: 2667–2679.
Rambaut A, Drummond AJ . (2007). Tracer v1.4. Available at http://beast.bio.ed.ac.uk/Tracer.
Raymond M, Rousset F . (1995). Genepop (version 1.2) population genetics software for exact tests and ecumenicism. J Hered 86: 248–249.
Rice WR . (1989). Analyzing tables of statistical tests. Evolution 43: 223–225.
Rodríguez-Cabello C, Sánchez F, Fernandez A, Olaso I . (2004). Is the lesser spotted dogfish (Scyliorhinus canicula from the Cantabrian Sea, a unique stock? Fish Res 69: 57–71.
Rogers AR, Harpending H . (1992). Population growth makes waves in the distribution of pairwise genetic differences. Mol Biol Evol 9: 552–569.
Sakellariou D, Lykousis V, Pousakis G . (2010). Reconstructing prehistoric landscapes in tectonically active regions: the Corinth and North Evia prehistoric lakes during LGM. XIX Congress of the Carpathian-Balkan Geological Association/Special Session S30, Thessaloniki, Greece, 23–26 September.
Schneider S, Excoffier L . (1999). Estimation of demographic parameters from the distribution of pairwise differences when the mutation rates vary among sites: Application to human mitochondrial DNA. Genetics 152: 1079–1089.
Schrey AW, Heist EJ . (2003). Microsatellite analysis of population structure in the shortfin mako (Isurus oxyrinchus. Can J Fish Aquat Sci 60: 670–675.
Stelbrink B, von Rintelen T, Cliff G, Kriwet J . (2010). Molecular systematics and global phylogeography of angel sharks (genus Squatina. Mol Phylogenet Evol 54: 395–404.
Stergiou KI, Christou ED, Georgopoylos D, Zenetos A, Souvermezoglou C . (1997). The Hellenic Seas: physics, chemistry, biology and fisheries. Oceanogr Mar Biol, Annu Rev 35: 415–538.
Takezaki N, Nei M, Tamura K . (2010). POPTREE2: software for constructing population trees from allele frequency data and computing other population statistics with windows interface. Mol Biol Evol 27: 747–752.
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley PF . (2004). MICROCHECKER: for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4: 535–538.
Verissimo A, McDowell JR, Graves JE . (2010). Global population structure of the spiny dogfish Squalus acanthias, a temperate shark with an antitropical distribution. Mol Ecol 19: 1651–1662.
Ward RD, Zemlak TS, Innes BH, Last PR, Hebert PDN . (2005). DNA barcoding Australia’s fish species. Philos T Roy Soc B 360: 1847–1857.
Wheeler A . (1978) Key to the Fishes of Northern Europe. Frederick Warne (Publishers) Ltd: London.
Whitney NM, Robbins WD, Schultz JK, Bowen BW, Holland KN . (2012). Oceanic dispersal in a sedentary reef shark (Triaenodon obesus: genetic evidence for extensive connectivity without a pelagic larval stage. J Biogeogr 39: 1144–1156.
Worm B, Davis B, Kettemer L, Ward-Paige CA, Chapman D, Heithaus MR et al. (2013). Global catches, exploitation rates and rebuilding options for sharks. Mar Policy 40: 194–204.
Acknowledgements
This study was conducted within the framework of the PhD thesis of Vasiliki Kousteni and has been co-financed by the European Union (European Social Fund-ESF) and Greek national funds through the Operational Programme ‘Education and Lifelong Learning’ of the National Strategic Reference Framework (NSRF)-Research Funding Programme: Heracleitus II: investing in a knowledge society through the European Social Fund. We deeply thank Dr Beatriz Guijarro and Dr Farid Hemida for collecting and providing the fin samples from the Balearic Islands and the Algerian Basin, respectively, as well as Dr Jon Bent Kristoffersen for suggestions on the data analysis. Three anonymous reviewers and the editor are also greatly acknowledged for their constructive comments in improving the quality of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Heredity website
Supplementary information
Rights and permissions
About this article

Cite this article
Kousteni, V., Kasapidis, P., Kotoulas, G. et al. Strong population genetic structure and contrasting demographic histories for the small-spotted catshark (Scyliorhinus canicula) in the Mediterranean Sea. Heredity 114, 333–343 (2015). https://doi.org/10.1038/hdy.2014.107
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2014.107
This article is cited by
-
Brown banded bamboo shark (Chiloscyllium punctatum) shows high genetic diversity and differentiation in Malaysian waters
Scientific Reports (2021)
-
Reviewing the genetic evidence for sex-biased dispersal in elasmobranchs
Reviews in Fish Biology and Fisheries (2021)
-
Strong genetic isolation despite wide distribution in a commercially exploited coastal shark
Hydrobiologia (2019)
-
The importance of considering genetic diversity in shark and ray conservation policies
Conservation Genetics (2018)
-
Phylogeography of the littoral prawn species Palaemon elegans (Crustacea: Caridea: Palaemonidae) across the Mediterranean Sea unveils disparate patterns of population genetic structure and demographic history in the two sympatric genetic types II and III
Marine Biodiversity (2018)
