
Abstract
The role of the immune system in neuropsychiatric diseases, including autism spectrum disorder (ASD), has long been hypothesized. This hypothesis has mainly been supported by family cohort studies and the immunological abnormalities found in ASD patients, but had limited findings in genetic association testing. Two cross-disorder genetic association tests were performed on the genome-wide data sets of ASD and six autoimmune disorders. In the polygenic score test, we examined whether ASD risk alleles with low effect sizes work collectively in specific autoimmune disorders and show significant association statistics. In the genetic variation score test, we tested whether allele-specific associations between ASD and autoimmune disorders can be found using nominally significant single-nucleotide polymorphisms. In both tests, we found that ASD is probabilistically linked to ankylosing spondylitis (AS) and multiple sclerosis (MS). Association coefficients showed that ASD and AS were positively associated, meaning that autism susceptibility alleles may have a similar collective effect in AS. The association coefficients were negative between ASD and MS. Significant associations between ASD and two autoimmune disorders were identified. This genetic association supports the idea that specific immunological abnormalities may underlie the etiology of autism, at least in a number of cases.
Similar content being viewed by others


Introduction
Autism spectrum disorder (ASD) is a broad spectrum of early-onset neuropsychiatric disorders characterized by severe deficits in social interaction and language, and the presence of repetitive and stereotyped behaviors and interests.1 Twin studies have demonstrated that ASD is largely genetic, with a 90% concordance between monozygotic twins, and heritable, with a 5–10 times higher familial risk than in the general population.2, 3 Genome-wide association studies (GWASs) have discovered significant genetic markers of single-nucleotide polymorphisms (SNPs)4, 5, 6 and de novo mutations of copy number variations7, 8, 9, 10 that may cause ASD. However, the molecular pathology of ASD is largely unclear owing to its genetic heterogeneity and the fact that only a small proportion of incidence is explained by known susceptibility loci.11, 12 Given this heterogeneity, it has been suggested that cross-disease analysis between ASD and other disorders that share common phenotypic symptoms or genetically susceptible loci will shed light on our understanding of the molecular mechanisms underlying ASD.13
The role of the immune system in neuropsychiatric diseases, including ASD, has long been hypothesized14, 15, 16 and is mainly supported by family cohort studies and the immunological abnormalities found in autistic patients. For example, Atladóttir et al.17 reported that the risk of ASD increases when a child's mother has rheumatoid arthritis or has a family history of type 1 diabetes. Other autoimmune disorders for which epidemiological studies have shown significant association with ASD, include maternal psoriasis,18 maternal ulcerative colitis,19 and autoimmune thyroid disease (ATD).20 In addition, various forms of immune dysregulation, including elevated cytokine levels21, 22, 23 and increased immunoglobulin and serum protein levels,24, 25, 26 have been identified in autistic children. However, few studies have reported potential genetic components that account for associations between ASD and autoimmune disorders. In fact, only three alleles, two in the human leukocyte antigen (HLA-DR4,27, 28 DR13,27 HLA-A229) and one in the major histocompatibility complex (MHC) of chromosome 6 (the C4B null allele30) have a confirmed association with ASD. With the advent of GWAS and the availability of large amounts of genotype data from both ASD and multiple autoimmune diseases, we now have the ability to directly analyze genetic associations across these diseases.
Materials and methods
Study samples and data quality control
We obtained Illumina BeadChip 550K genotype data of 941 multiplex families (Illumina Inc., San Diego, CA, USA) with autistic children from the Autism Genetic Resource Exchange (AGRE).31 For the phenotype labeling, we followed the classification of the Autism Diagnostic Interview—Revised32 included in the AGRE phenotype database, but excluded individuals whose Autism Diagnostic Interview—Revised classification and Autism Diagnostic Observation Schedule33 classification did not agree. We examined each family's data to find monozygotic twins, triplets, or quadruplets and elected to include only one monozygotic sibling per family, filtering 72 individuals from the data set. We also removed 92 individuals who were annotated as ‘possible non-idiopathic autism’ in the AGRE phenotype database. These cases include prematurity of less than 35 weeks gestation (45 individuals), Fragile X syndrome (12 individuals), known chromosomal abnormality (10 individuals), and other diagnosed neurogenetic disorders (10 individuals). We further applied a set of quality control filters in order to identify a stringent subset of robust SNPs. We excluded 32 individuals with genotyping rate <0.95 and 18510 SNPs with genotyping rate <0.95. We examined Mendelian error per each family trio, and removed seven individuals for Mendelian error >1 percent of all markers. SNPs with a minor allele frequency <0.05 or with a Hardy–Weinberg equilibrium exact test P<0.001 were excluded from further analysis. After quality control, the ASD genomic data consisted of 1397 affected trios and a total of 470025 SNPs. We used the software package PLINK34 to conduct the transmission disequilibrium test (TDT) with all family trios passing quality control without identifying subpopulation, as the TDT is known to maintain the desired type 1 error rate in the presence of population stratification.35
We obtained two groups of GWAS data from the Wellcome Trust Case Control Consortium (WTCCC) as target disease sets for measuring association with ASD. The smaller set (WTCCC4) consisted of 1500 common controls and 1000 independent cases of ankylosing spondylitis (AS), autoimmune thyroid disease, multiple sclerosis (MS), and breast cancer. It had 14436 non-synonymous SNPs plus 897 SNPs in the major histocompatibility complex region, and was genotyped using the Illumina Infinium 15K array. The larger set (WTCCC7) in the Affymetrix GeneChip 500K array (Affymetrix Inc., Santa Clara, CA, USA) consisted of 3000 shared controls and 2000 independent cases in seven diseases, including bipolar disorder, coronary artery disease, Crohn's disease, hypertension, rheumatoid arthritis, type 1 diabetes and type 2 diabetes. Shared control samples came from two sources: for the smaller set (WTCCC4), control samples were taken from the 1958 British Birth Cohort (58C) and for the larger set (WTCCC7), 1500 control samples were taken from 58C. The remaining 1500 samples were taken from blood donors recruited by the three UK Blood Services (UKBS).36, 37 Although the ages of the former are known and past the typical age of onset for the autoimmune conditions studied, the individual age information of the UKBS group is not known. The potential variation in ages from this control group could introduce minor classification bias, as some of individuals may (or may have) develop the autoimmune disorder in the future, as discussed in the main WTCCC7 article.37 However, the prevalence of these autoimmune disorders is relatively rare, with a combined prevalence of rate of up to 8 percent.38 Therefore, although we do not have detailed age information for the UKBS samples, we assume that misclassification due to age of onset would be rare and unlikely to bias our results. We followed the quality control steps described in Burton et al.,36, 37 and further removed SNPs with minor allele frequency <0.05 or Hardy–Weinberg equilibrium exact test P<0.001. After quality control steps, there were 12700 SNPs remaining in the WTCCC4 set, and 469557 SNPs were retained for further analysis in the WTCCC7 set. Across all SNPs passing quality control, we used case–control association analysis with the software package PLINK.
As these data were genotyped using different platforms, we cross-referenced SNP ids and strand information as follows. First we converted all custom, non-reference SNP ids into corresponding reference SNP ids by querying the UCSC Genome Browser (http://www.genome.uscs.edu; version May 2004/NCBI genome build 35). Then we examined all reference SNP ids to confirm that they are up-to-date by querying NCBI dbSNP (build 132) and removed six SNPs that had multiple identifiers. Next, we checked all the allelic and chromosomal position information of each data set with the HapMap genotype data (CEU founders, release 23), reconfiguring the strands when alleles did not match. Mapping SNPs into the corresponding gene regions was done by querying NCBI dbSNP (build 132). The number of intersection SNPs between ASD and WTCCC4 set was 5318, and the number of intersection SNPs between ASD and WTCCC7 set was 73331.
Polygenic score analysis
To identify genomic associations between diseases, we used two separate and complementary approaches, as illustrated in Figure 1. The first, the polygenic score (PS) test39 measured the collective effect of disease-associated SNPs from one disease on a collection of SNPs from another. The test was designed to identify associations between complex multigenic diseases that manifest through a combination of multiple variants of small individual effect. We used the PS test here to determine if collections of variants from cases with ASD appear to correlate with groups of variants from individuals with autoimmune disorders. First, we labeled a GWAS data set from one disease as the ‘source’ data set and another GWAS from a different disease as the ‘target’. From the source data set we selected groups of autosomal SNPs that corresponded to a range of nominal significance thresholds (PT) as source alleles, and recorded the minor allele and odds ratio information of those selected SNPs. Then, for each individual in the target data set, we calculated the PS by computing the average number of source alleles that the individual had, weighted by the logarithm-of-the-odds-ratio (log (odds-ratio)) from the source data set. This polygenic score can be considered to be a measure of probabilistic similarity for those SNPs in the source disease data set and each individual in the target data set. As such, if the source and target diseases were highly similar in allelic composition, the cases would yield consistently higher scores than the controls. If there were multiple target diseases, ones that were more closely related to the source disease would yield a higher average PS, as demonstrated in International Schizophrenia Consortium.39 To provide this context, we ran logistic regression analysis with the PS to predict the classification of the target disease. Then, we estimated the variance explained in the target disease data by the PS using the Nagelkerke's pseudo R-square40 from a model with the PS and covariates, versus that from a model without the score. We took the total number of alleles used to calculate the PS and the numerically coded site information of individuals in the WTCCC data sets as covariates.

Analysis steps in the polygenic score approach (a) and genetic variation score approach (b). In both approaches, P-values and odd-ratios were calculated from transmission disequilibrium test (ASD data) or case/control association test (WTCCC data) after quality control steps. (a) From the ASD data set and a given P value threshold PT, a set of SNPs with P<PT were selected as ‘source alleles’. Then for each target disease data set, the polygenic score was calculated for each individual. We conducted logistic regression analysis with the polygenic scores in order to examine whether source alleles from ASD data could explain variances in the target disease data sets. (b) For each ASD and autoimmune data set, the genetic variation score was calculated for each SNP. We used Pearson's pair-wise correlation to compare the collective effect of SNPs in two diseases.
Genetic variation score analysis
The second metric we used to compare ASD to autoimmune disorders was the genetic variance score (GVS).39 With GVS, a combined score of the odds-ratio and P-value was defined for each disease data set and every SNP belonging to that set. Specifically, for each disease data set d and a SNP s∈d, a GVS of [d, s] was defined as sign (log(odds-ratio [d, s])) * (−log10(P-value [d, s])). Given that the odds-ratio was calculated with respect to the minor allele of the SNP and that the odds-ratio was greater than 1 when the minor allele was more likely to occur in the case group, the sign (log (odds-ratio)) was positive when the minor allele was the risk allele, and negative when the major allele was the risk allele. When the GVS scores of a SNP in two diseases had different signs, we assumed that the allele was protective in one disease and deleterious in the other. As an extension of this allele-specific comparison, we computed the Pearson pair-wise correlation between the GVS data of two diseases. This correlation analysis enabled us to determine whether large numbers of SNPs have similar effects in two different diseases, that is, a high positive correlation coefficient indicated similar effects, whereas a strong negative coefficient indicated opposing effects of the risk alleles.
Although both of these two approaches were based on individual SNP association and framed in terms of odds-ratios and P-values, they provided two complementary assessments of the association between the genotypes of two different diseases. The PS test assigned a score per individual in the target disease set and assumed that many weakly associated SNPs with marginal odds-ratios may work collectively, such that they have stronger association test statistics than loci drawn from the null distribution. The GVS and its Pearson correlation as a similarity measure examined whether there were SNPs with strong significance in both disease sets, as GVS compared the pair-wise, allele-specific significance of each SNP.
Results
PS analysis
We examined whether groups of ASD-associated SNPs can collectively account for genomic variation in another disease, even when each individual SNP may not have a very strong effect. From this source sample, we selected sets of marginally associated alleles at five different P-value thresholds (PT <0.01, 0.05, 0.1, 0.25 and 0.5). After calculating PS per each individual in each target autoimmune data set, we performed a logistic regression analysis using the PS as a predictor of target disease classification, and estimated the variance in the target sample explained by the PS using Nagelkerke's pseudo R2. Table 1 summarizes this R2 measure with P-value thresholds for each group of source alleles and target diseases. Source alleles from ASD data showed a significant enrichment in AS cases (P=2.22 × 10−16 at PT<0.5), explaining about two percent of variance. In addition, the effect of source alleles became larger when the P-value threshold PT was increased, supporting our hypothesis that alleles with little individual effect in one disease may work collectively in other diseases. In MS, source alleles showed a similar pattern of increasing R2 with increasing PT thresholds, explaining about one percent of variance (P=5.16 × 10−5 at PT < 0.5). In contrast to AS, however, the coefficients of the PS in MS were all negative and the average mean score was higher in controls (data not shown), suggesting that ASD risk alleles have opposite overall effects in these two diseases. In other WTCCC target diseases, the variance explained by ASD source alleles was either too small (R2 < 0.01), or was not significant (P>0.05) over all P-value thresholds.
Genetic variation score analysis
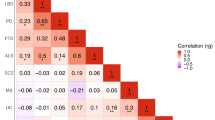
Next, we examined whether allele-specific associations can be found among ASD and autoimmune disease data sets via the GVS approach. As we examined the collective effect of marginally significant SNPs and found enrichment in two autoimmune diseases in the PS approach, here we focused our analysis on SNPs with at least nominally significant P values (P < 0.05). Multiple hypothesis correction was not applied to the P values when calculating GVS because we examined associations of disease pairs by correlation coefficients, which would not change after multiple hypothesis correction. Table 2 shows the Pearson correlation coefficients for all disease pairs within ASD and the WTCCC data sets, calculated with GVS. Consistent with the results from the PS analysis above, the ASD data exhibited significant positive association with AS (coefficient 0.4032) and significant negative association with MS (coefficient −0.3092). AS and MS were also strongly negatively associated with one another (coefficient −0.3092). Interestingly, the coefficient of association between ASD and AS was higher than any of the autoimmune disease pairs, while the strength of the association between ASD and MS was comparable to that of strengths of association between any pair of autoimmune disorders. Autoimmune thyroid disease was slightly positively correlated with ASD (coefficient 0.2012), whereas all other autoimmune diseases (MS, Crohn's disease and rheumatoid arthritis) showed little association with ASD in their profiles. Table 3 summarizes the nominally significant SNPs in both ASD and the autoimmune diseases, including AS and MS. Many of these SNPs fell within already known ASD risk genes (for example, rs2034648—AGAP1) or mental disorder susceptibility genes (for example, rs11643718–SLC12A3 and rs3132468–MICB). Another group of SNPs fell within the protein coding regions of several known autoimmune risk genes (for example, rs3130559–PSORS1C1, rs3129943–C6orf10 and rs3130542–HLA-C). However, none of these have been reported previously as ASD susceptibility genes.
Discussion
Given that the magnitude of the association between ASD and the two autoimmune diseases, AS and MS, was either greater than or on par with the strength of association between what are considered now to be genetically similar autoimmune diseases, coupled with the lack of any other association of the same significance between ASD and the remaining autoimmune disorders examined, our results clearly demonstrated that there are true genomic links between ASD and the two autoimmune diseases, links that likely can inform our understanding of the genetics and treatments of ASD. However, further study and verification is required to characterize and explain these particular genomic associations. An interesting, albeit anecdotal, similarity between ASD and AS is that they both have an appreciable male bias. AS has a male-to-female ratio of approximately 2.5:142 that is of the same magnitude as the male bias of ASD's 4:143 while other autoimmune diseases, most importantly including MS,42, 43 generally show higher susceptibility in females. Also of interest to the observed genomic similarities between ASD and AS is anti-TNF (tumor necrosis factor) alpha treatment therapy. Anti-TNF agents are among the most efficacious options for treatment of AS,46, 47 and, although not well studied to date, ASD cases have been shown to have increased expression levels of TNF-alpha and IL-6,48 and separate cases have received anti-TNF therapy.49 Interestingly, anti-TNF agents tend to cause demyelination as an adverse side effect,50 a symptom that is typical in MS.
To rule out the possibility that the ratio of males to females in our original data sets biased our findings, we constructed sex-balanced and gender-specific data sets and recomputed the polygenic scores. The direction and relative strength of correlation with AS and MS remained unchanged, indicating that our results are not unduly influenced by the different numbers of male and female cases in the data sets. To rule out the possibility that differences in population heterogeneity and ethnic background between the ASD and the autoimmunity data collections biased the findings, we tested for association using only ASD individuals of European ancestry to match the ancestry of the individuals included within the autoimmune data sets. With a reduced number of 1019 affected trios from the ASD data set, we conducted TDT analysis and PS testing and found results consistent with those found using the complete ASD data set, a positive association with AS and negative association with MS and no other significant associations between ASD and the remaining autoimmune disorders studied. These results suggest that differences in mixed ancestry between the two data sets did not influence the associations discovered. However, this lack of bias from mixed ancestry does not preclude the possibility that geographical differences (our ASD collection was from the United States, while autoimmune collections were from the United Kingdom), and consequent differences in environmental exposure, could influence risk for disease differently as has been shown in the MS cases.51 However, differences in exposure related to geography are more likely to cause underestimates of association rather than overestimates and are thus unlikely to alter the results shown here. An additional potential bias could arise from the age of onset of autoimmune conditions. For example, MS tends to manifest in women during childbearing ages. Our ASD sample contains mothers who have, by definition, passed childbearing age, but because the mothers served as controls in the association testing, the observed negative association between ASD and MS suggests that mothers in the ASD set had a higher loading of MS-related alleles, not a lower loading. Under this assumption, it is unlikely that differences in ages of the sampled populations biased the significance or directionality of the association identified. Nevertheless, further studies with more equivalent samples across parameters, including age, ancestry and geography would be valuable to verify our findings.
In conclusion, we found significant, allele-specific genomic associations between ASD and two autoimmune diseases, AS and MS, which were supported using two complementary analytical strategies. The first, a PS approach, revealed that a collection of relatively weakly associated ASD susceptibility markers could still explain a significant percentage of the variation in both AS and MS cases. Coefficients from logistic regression analysis with the polygenic scores showed that the collective, allele-specific role of SNPs in ASD and AS was similar, whereas the roles of the alleles explaining the similarity between ASD and MS were of opposite effect, conferring risk in one and protection from onset in the other. The second, a genetic variation score approach, found the same results of allele-specific association between ASD and AS and ASD and MS with comparable or higher strength of association than that found between any autoimmune disease pair. Together these results suggest that common genetic mechanisms exist between ASD and AS and that opposing genetic mechanisms exist between ASD and MS. Both approaches pinpoint sets of SNPs that comprise the significant associations seen in our study and that may be of value as targets for further experiments aimed at understanding the genetic ties between ASD and autoimmune diseases.
References
Fombonne E . Epidemiology of pervasive developmental disorders. Pediatr Res 2009; 65: 591–598.
Bailey A, Le Couteur A, Gottesman I, Bolton P, Simonoff E, Yuzda E et al. Autism as a strongly genetic disorder: evidence from a British twin study. Psychol Med 1995; 25: 63–77.
Ronald A, Hoekstra RA . Autism spectrum disorders and autistic traits: a decade of new twin studies. Am J Med Genet B Neuropsychiatr Genet 2011; 156: 255–274.
Wang K, Zhang H, Ma D, Bucan M, Glessner JT, Abrahams BS et al. Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature 2009; 459: 528–533.
Weiss LA, Arking DE, Daly MJ, Chakravarti A, Arking DE, Brune CW et al. A genome-wide linkage and association scan reveals novel loci for autism. Nature 2009; 461: 802–808.
Anney R, Klei L, Pinto D, Regan R, Conroy J, Magalhaes TR et al. A genome-wide scan for common alleles affecting risk for autism. Hum Mol Genet 2010; 19: 4072–4082.
Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T et al. Strong association of de novo copy number mutations with autism. Science 2007; 316: 445–449.
Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, Fossdal R et al. Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med 2008; 358: 667–675.
Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, Wood S et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 2009; 459: 569–573.
Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature 2010; 466: 368–372.
Folstein SE, Rosen-Sheidley B . Genetics of autism: complex aetiology for a heterogeneous disorder. Nat Rev Genet 2001; 2: 943–955.
Geschwind DH . Advances in autism. Annu Rev Med 2009; 60: 367–380.
Wall DP, Esteban FJ, Deluca TF, Huyck M, Monaghan T, Velez de Mendizabal N et al. Comparative analysis of neurological disorders focuses genome-wide search for autism genes. Genomics 2009; 93: 120–129.
Money J, Bobrow NA, Clarke FC . Autism and autoimmune disease: a family study. J Autism Child Schizophr 1971; 1: 146–160.
Licinio J, Wong ML . The role of inflammatory mediators in the biology of major depression: central nervous system cytokines modulate the biological substrate of depressive symptoms, regulate stress-responsive systems, and contribute to neurotoxicity and neuroprotection. Mol Psychiatry 1999; 4: 317–327.
Korvatska E, Van de Water J, Anders TF, Gershwin ME . Genetic and immunologic considerations in autism. Neurobiol Dis 2002; 9: 107–125.
Atladóttir HO, Pedersen MG, Thorsen P, Mortensen PB, Deleuran B, Eaton WW et al. Association of family history of autoimmune diseases and autism spectrum disorders. Pediatrics 2009; 124: 687–694.
Croen LA, Grether JK, Yoshida CK, Odouli R, Van de Water J . Maternal autoimmune diseases, asthma and allergies, and childhood autism spectrum disorders. a case-control study. Arch Pediatr Adolesc Med 2005; 159: 151–157.
Mouridsen SE, Rich B, Isager T, Nedergaard NJ . Autoimmune diseases in parents of children with infantile autism: a case-control study. Dev Med Child Neurol 2007; 49: 429–432.
Sweeten TL, Bowyer SL, Posey DJ, Halberstadt GM, McDougle CJ . Increased prevalence of familial autoimmunity in probands with pervasive developmental disorders. Pediatrics 2003; 112: e420.
Molloy CA, Morrow AL, Meinzen-Derr J, Schleifer K, Dienger K, Manning-Courtney P et al. Elevated cytokine levels in children with autism spectrum disorder. J Neuroimmunol 2006; 172: 198–205.
Li X, Chauhan A, Sheikh AM, Patil S, Chauhan V, Li XM et al. Elevated immune response in the brain of autistic patients. J Neuroimmunol 2009; 207: 111–116.
Ashwood P, Krakowiak P, Hertz-Picciotto I, Hansen R, Pessah I, Van de Water J . Elevated plasma cytokines in autism spectrum disorders provide evidence of immune dysfunction and are associated with impaired behavioral outcome. Brain Behav Immun 2011; 25: 40–45.
Croonenberghs J, Wauters A, Devreese K, Verkerk R, Scharpe S, Bosmans E et al. Increased serum albumin, gamma globulin, immunoglobulin IgG, and IgG2 and IgG4 in autism. Psychol Med 2002; 32: 1457–1463.
Enstrom A, Krakowiak P, Onore C, Pessah IN, Hertz-Picciotto I, Hansen RL et al. Increased IgG4 levels in children with autism disorder. Brain Behav Immun 2009; 23: 389–395.
Angelidou A, Francis K, Vasiadi M, Alysandratos KD, Zhang B, Theoharides A et al. Neurotensin is increased in serum of young children with autistic disorder. J Neuroinflammation 2010; 7: 48.
Torres AR, Maciulis A, Stubbs EG, Cutler A, Odell D . The transmission disequilibrium test suggests that HLA-DR4 and DR13 are linked to autism spectrum disorder. Hum Immunol 2002; 63: 311–316.
Johnson WG, Buyske S, Mars AE, Sreenath M, Stenroos ES, Williams TA et al. HLA-DR4 as a risk allele for autism acting in mothers of probands possibly during pregnancy. Arch Pediatr Adolesc Med 2009; 163: 542–546.
Torres AR, Sweeten TL, Cutler A, Bedke BJ, Fillmore M, Stubbs EG et al. The association and linkage of the HLA-A2 class I allele with autism. Hum Immunol 2006; 67: 346–351.
Odell D, Maciulis A, Cutler A, Warren L, McMahon WM, Coon H et al. Confirmation of the association of the C4B null allele in autism. Hum Immunol 2005; 66: 140–145.
Lajonchere CM, Brown WT, Bucan M, Cantor R, Constantino J, Geschwind D et al. Changing the landscape of autism research: the autism genetic resource exchange. Neuron 2010; 68: 187–191.
Lord C, Rutter M, Le Couteur A . Autism Diagnostic Interview-Revised: a revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J Autism Dev Disord 1994; 24: 659–685.
Lord C, Rutter M, Goode S, Heemsbergen J, Jordan H, Mawhood L et al. Autism diagnostic observation schedule: a standardized observation of communicative and social behavior. J Autism Dev Disord 1989; 19: 185–212.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007; 81: 559–575.
Ewens WJ, Spielman RS . The transmission/disequilibrium test. history, subdivision, and admixture. Am J Hum Genet 1995; 57: 455–464.
Burton PR, Clayton DG, Cardon LR, Craddock N, Deloukas P, Duncanson A et al. Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat Genet 2007; 39: 1329–1337.
Burton PR, Clayton DG, Cardon LR, Craddock N, Deloukas P, Duncanson A et al. Genome-wide association study of 14 000 cases of seven common diseases and 3000 shared controls. Nature 2007; 447: 661–678.
International Schizophrenia Consortium, Purcell SM, Wray NR, Stone JL, Visscher PM, O'Donovan MC et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 2009; 460: 748–752.
National Institutes of Health, The Autoimmune Diseases Coordinating Committee. Progress in autoimmune diseases research. 2005; http://www.niaid.nih.gov/topics/autoimmune/Documents/adccfinal.pdf.
Nagelkerke N . A note on a general definition of the coefficient of determination. Biometrika 1991; 78: 691.
Sirota M, Schaub MA, Batzoglou S, Robinson WH, Butte AJ . Autoimmune disease classification by inverse association with SNP alleles. PLoS Genet 2009; 5: e1000792.
Calin A, Brophy S, Blake D . Impact of sex on inheritance of ankylosing spondylitis: a cohort study. Lancet 1999; 354: 1687–1690.
Newschaffer CJ, Croen LA, Daniels J, Giarelli E, Grether JK, Levy SE . et al. The epidemiology of autism spectrum disorders. Annu Rev Public Health 2007; 28: 235–258.
Compston A, Coles A . Multiple sclerosis. Lancet 2008; 372: 1502–1517.
Alonso A, Hernan MA . Temporal trends in the incidence of multiple sclerosis. a systematic review. Neurology 2008; 71: 129–135.
Kalden JR . Emerging role of anti-tumor necrosis factor therapy in rheumatic diseases. Arthritis Res 2002; 4 (Suppl 2): 34–40.
Silva LC, Ortigosa LC, Benard G . Anti-TNF agents in the treatment of immune-mediated inflammatory diseases: mechanisms of action and pitfalls. Immunotherapy 2010; 2: 817–833.
Malik M, Sheikh AM, Wen G, Spivack W, Brown WT, Li X . Expression of inflammatory cytokines, Bcl2 and cathepsin D are altered in lymphoblasts of autistic subjects. Immunobiology 2011; 216: 80–85.
Chez M, Guido-Estrada M . Immune therapy in autism: historical experience and future directions with immunomodulatory therapy. Neurotherapeutics 2010; 7: 299–301.
Davis SA, Johnson RR, Pendleton JW . Demyelinating disease associated with use of etanercept in patients with seronegative spondyloarthropathies. J Rheumatol 2008; 35: 1469–1470.
Simpson Jr S, Blizzard L, Otahal P, Van der Mei I, Taylor B . Latitude is significantly associated with the prevalence of multiple sclerosis: a meta-analysis. J Neurol Neurosurg Psychiatry 2011; 82: 1132–1141.
Acknowledgements
We thank Drs Vincent Fusaro, Jike Cui and Todd F DeLuca for their helpful discussions while conducting the analyses. We also thank the anonymous reviewers for their insightful reviews that strengthened our manuscript. This work was supported by the National Institutes of Health under Grant No. 1R01MH090611-01A1 awarded to DPW.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Jung, JY., Kohane, I. & Wall, D. Identification of autoimmune gene signatures in autism. Transl Psychiatry 1, e63 (2011). https://doi.org/10.1038/tp.2011.62
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tp.2011.62
Keywords
This article is cited by
-
Association Analysis of ANRIL Polymorphisms and Haplotypes with Autism Spectrum Disorders
Journal of Molecular Neuroscience (2021)
-
The putative role of environmental aluminium in the development of chronic neuropathology in adults and children. How strong is the evidence and what could be the mechanisms involved?
Metabolic Brain Disease (2017)
-
Gene expression analysis in Fmr1KO mice identifies an immunological signature in brain tissue and mGluR5-related signaling in primary neuronal cultures
Molecular Autism (2015)
