
Abstract
Intracellular drug exposure is influenced by cell- and tissue-dependent expression of drug-transporting proteins and metabolizing enzymes. Here, we introduce the concept of intracellular bioavailability (Fic) as the fraction of extracellular drug available to bind intracellular targets, and we assess how Fic is affected by cellular drug disposition processes. We first investigated the impact of two essential drug transporters separately, one influx transporter (OATP1B1; SLCO1B1) and one efflux transporter (P-gp; ABCB1), in cells overexpressing these proteins. We showed that OATP1B1 increased Fic of its substrates, while P-gp decreased Fic. We then investigated the impact of the concerted action of multiple transporters and metabolizing enzymes in freshly-isolated human hepatocytes in culture configurations with different levels of expression and activity of these proteins. We observed that Fic was up to 35-fold lower in the configuration with high expression of drug-eliminating transporters and enzymes. We conclude that Fic provides a measurement of the net impact of all cellular drug disposition processes on intracellular bioavailable drug levels. Importantly, no prior knowledge of the involved drug distribution pathways is required, allowing for high-throughput determination of drug access to intracellular targets in highly defined cell systems (e.g., single-transporter transfectants) or in complex ones (including primary human cells).
Similar content being viewed by others
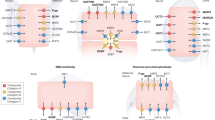


Introduction
Drug-transporting proteins and metabolizing enzymes are determinants of intracellular drug disposition1,2: uptake transporters increase the drug concentration in the cell interior3,4,5, while efflux transporters6,7,8 and enzymes9,10 reduce the amount of intracellularly available compound (Fig. 1). Therefore, these proteins are of major importance for drug efficacy and toxicity11. The contribution of such drug-disposition proteins has been studied in vitro, e.g., in cell lines expressing specific transporters5,12,13,14,15, in hepatocytes (in suspension, monolayer and sandwich cultures)16,17, and in more complex systems, such as tissue slices18 and 3D cultures19. Typically, only the relative impact of transporters on the intracellular compound concentration is reported, i.e., fold-differences in intracellular concentration when the transporter or metabolic enzymes are active (or overexpressed) compared to when these proteins are inhibited (or absent). Quantitative determination of the intracellular unbound drug concentration, which is the relevant concentration for binding to intracellular target proteins, either relies on experimentally demanding setups in freshly isolated cells3,4,5,20,21 or tissues22,23, or on a priori knowledge of transport and metabolic mechanisms for the compound being studied24. To overcome these limitations, we recently developed a simple methodology to measure intracellular unbound drug concentrations in cultured cells25, compatible with high-throughput formats26,27. We also showed that our methodology correlates with binding to an intracellular target (thymidylate synthase)28.

Uptake transporters (green, e.g. OATP1B1) increase the Fic, while efflux transporters (light orange, e.g. P-gp) and metabolizing enzymes (dark orange, e.g. CYP3A4) lower the Fic. Arrows represent diffusion over the plasma membrane (passive lipoidal transmembrane diffusion or carrier-mediated diffusion).
In this work, we apply this methodology to study the impact of drug-transporting proteins and metabolizing enzymes on intracellular unbound compound concentrations. For this purpose, we introduced the concept of intracellular compound bioavailability (Fic), which is the fraction of the externally added compound concentration that is available to bind targets in the cell interior. Throughout this study, this concept is equivalent to the unbound drug accumulation ratio (Kpuu), a term commonly used in pharmacokinetic studies of blood-to-tissue concentration ratios22. However, there are situations in which the Kpuu term is not applicable (e.g., when experiments are performed in the presence of serum proteins). First, we evaluated how Fic is affected by a single uptake or a single efflux transporter. We used well defined cell lines overexpressing the organic anion-transporting polypeptide 1B1 (OATP1B1; SLCO1B1*1a) and P-glycoprotein (P-gp; ABCB1), two of the transporters most commonly involved in drug disposition1. We then studied the parallel impact of multiple transporters and enzymes in a more complex system, freshly isolated human hepatocytes. The hepatocytes were used in two configurations known to display different gene expression and activity of transporters and enzymes: directly after isolation and after 24 h of culture in monolayer format29,30,31. In conclusion, our simple methodology provides a good estimate of compound available to bind targets in the cell interior, even in complex cell systems.
Results
Impact of OATP1B1 on intracellular bioavailability (Fic) of compounds
We investigated the impact of drug-transporting proteins on intracellular compound bioavailability (Fic). First, we studied the influence of an important uptake transporter (Fig. 1) by using HEK293 cells transfected with human OATP1B1 or with an empty vector (mock-transfected cells; Fig. 2a). We measured the intracellular fraction of unbound compound (fu,cell) in cell homogenates and cellular compound accumulation (Kp) in live cells (see ‘Methods’ for details). From these parallel experiments, Fic was then calculated as the product of fu,cell and Kp, providing a measure of how much of an extracellularly applied drug concentration is freely available in the cell interior.

(a) Schematic representation of cell types used in this study: mock-transfected HEK293 cells express negligible levels of relevant drug-transporting proteins; OATP1B1-transfected HEK293 cells express OATP1B1. (b) Comparison between fu,cell in mock-transfected and OATP1B1-transfected HEK293 cells. Shaded area represents the impact of a theoretical error on fu,cell from measurements of fu,hom with 15% error. (c) Comparison between Fic in mock-transfected and OATP1B1-transfected HEK293 cells at 0.1 μM compound concentration. In b and c, negatively charged compounds at pH 7.4 are represented as triangles, neutral and zwitterionic species are represented by circles, and positively charged compounds are represented by squares. Substrates of OATP1B1 are highlighted in green. (d–g) Concentration-dependence of Fic in OATP1B1-transfected HEK293 cells (green filled squares), fitted with a sigmoidal model (green line), and mock-transfected HEK293 cells (blue circles) for atorvastatin (d), pitavastatin (e), fluvastatin (f), and simvastatin acid (g). For simvastatin acid, intracellular concentrations at 0.01 μM were below limit of quantification.
Measurements of fu,cell showed no significant difference between mock- and OATP1B1-transfected cells (Fig. 2b; Supplementary Table 2; rS = 0.97). This indicates a negligible contribution from OATP1B1 to the total binding to cellular proteins, which can be explained by previous observations that a non-saturable component, most likely the membrane lipids, is the major cellular binding site25,32,33,34. Further, OATP1B1 contributes only 0.03% of all protein in the transfected cells (12 pmol/mg membrane protein35, assuming membrane proteins to be 30% of the total cellular protein mass36).
In contrast, substrates of OATP1B1 showed higher total cellular accumulation (Kp) in OATP1B1- than in mock-transfected cells (Supplementary Table 2). This difference was also reflected in Fic, which was, on average, 2.9-fold higher in OATP1B1-transfected cells (range: 1.1–9.8-fold; p = 0.001 in Wilcoxon matched-pairs signed rank test) (Fig. 2c). In contrast, non-substrates of OATP1B1 showed similar Fic in both cell lines (range: 0.57–1.9-fold difference between OATP1B1- and mock-transfected cells; p = 0.06 in Wilcoxon matched-pairs signed rank test). Atorvastatin and pitavastatin displayed the largest increase in Fic from mock-transfected cells (9.8 and 8.3-fold higher in OATP1B1-transfected cells, respectively). This is in agreement with that OATP transport is the rate-limiting step in the liver disposition of these statins12,13,37,38. The impact of the uptake transporter decreased with increasing lipophilicity of the compounds (rS = −0.63, p = 0.04 for the rank-order correlation between difference in Fic and log D of substrates; Supplementary Figure 1b). This most likely reflects the well-established increase in transmembrane diffusion with increasing lipophilicity39,40,41.
To further investigate if the observed differences were caused by OATP1B1-mediated uptake, we measured Fic for four substrates (atorvastatin, pitavastatin, fluvastatin, and simvastatin acid) across a range of concentrations (Fig. 2d–g). In mock-transfected HEK293 cells, which have negligible basal expression of drug-transporting proteins42, Fic was independent of compound concentration for all substrates, indicating that passive permeability across the cell membrane was the main uptake mechanism in these cells. In contrast, in OATP1B1-transfected cells, Fic decreased with increasing compound concentration, approaching levels in mock-transfected cells at higher concentrations. The concentrations at which Fic was half of its maximal value were close to previously reported Km values for OATP1B1-mediated transport of the respective substrates12,13,43, reflecting saturation of the transporter and an ensuing dominance of a passive mechanism.
Impact of P-gp on intracellular bioavailability (Fic) of compounds
We next evaluated how efflux transporters affect Fic (Fig. 1) through measurements in MDCK cells transfected with human P-gp. To determine baseline Fic in these cells in the absence of P-gp, we used wild-type MDCK cells where background endogenous canine P-gp was knocked out using CRISPR-Cas9 technology (cP-gp-KO)44 (Fig. 3a)—see ‘Supplementary results’ for a discussion on the choice of these cells as a control.

(a) Schematic representation of cell types used in this study: MDCK cells where canine P-gp has been knocked-out using CRISPR/Cas 9 (cP-gp-KO); MDCK cells transfected with human P-gp, expressing both canine and human P-gp. (b) Comparison between Fic in cP-gp-KO and in P-gp-transfected MDCK cells at 0.5 μM compound concentration. Negatively charged compounds at pH 7.4 are represented as triangles, neutral and zwitterionic species are represented by circles, and positively charged compounds are represented by squares. Substrates of P-gp are highlighted in yellow. Nelfinavir is represented in parentheses as its Fic is outside the range of this plot in P-gp-transfected cells. (c) Relationship between simvastatin acid concentration and Fic in P-gp-transfected MDCK cells (yellow filled squares), P-gp-KO MDCK cells (blue circles), and P-gp-KO MDCK cells pre-incubated (15 min) followed by co-incubation with 10 μM cyclosporine A (gray triangles). (d) Kinetic cell model simulations that best described experimental observations of concentration-dependence Fic for simvastatin acid (conditions: passive permeability = 10 × 10−6 cm/s; Vmax,uptake = 1000 pmol/min/mg protein; Km,uptake = 10 μM; Vmax,efflux = 1000 pmol/min/mg protein; Km,efflux = 100 μM).
As in the experiments with HEK293 cells, we first measured fu,cell in MDCK cells transfected with human P-gp (Supplementary Table 3). We used this value of fu,cell for all MDCK cell types used in this study, based on our observation above that the low levels at which transporters are expressed (compared to total protein) do not affect binding (Fig. 2b; P-gp contributes only 0.1% of all protein in P-gp-transfected MDCK cells (6.0 pmol/mg total protein, in-house data)).
We then combined fu,cell and Kp measurements to calculate Fic in P-gp-transfected MDCK cells and in cP-gp-KO cells (Supplementary Table 3). Because of the active efflux of substrates when the transporter is present, the Fic of compounds previously reported to be P-gp substrates was on average 2.0-fold lower in P-gp-expressing than in cP-gp-KO cells (range: 0.94–20-fold lower in P-gp transfected than cP-gp-KO cells; p < 0.001 in Wilcoxon matched-pairs signed rank test; Fig. 3b). For compounds not previously described as substrates, Fic was more similar in the two cell lines (range: 1.1–3.0-fold difference between cP-gp-KO cells and P-gp transfected cells), but significantly different (p = 0.001; see discussion below). Nelfinavir, an established P-gp substrate, displayed a remarkably low Fic in P-gp-transfected cells (20-fold lower than in cells not expressing P-gp). This indicates that P-gp contributes strongly to the cellular elimination of nelfinavir, in particular in cells that do not metabolize this compound. Overall, lipophilic molecules showed lower Fic in P-gp transfected cells (rS = −0.36, p = 0.05 for the rank-order correlation between difference in Fic and log D of all compounds; Supplementary Figure 2b), independently of being previously described as substrates or not. This possibly reflects that compounds bind P-gp from within the inner leaflet of the plasma membrane45,46, and suggests that additional compounds in our dataset may be substrates of P-gp, besides those previously reported as such.
As for the OATP1B1 substrates, we measured the concentration-dependence of Fic in P-gp-expressing and cP-gp-KO cells. For simvastatin acid, Fic in P-gp-expressing cells was lower than that in cP-gp-KO cells at low concentrations and approached it at higher concentrations (Fig. 3c). Surprisingly, Fic decreased with increasing compound concentration in both the P-gp expressing cells and in the cP-gp-KO cells, which was not caused by a saturation of binding sites25 (Supplementary Figure 4). Similar results were observed for loperamide (Supplementary Figure 3). When inhibiting active transport with 10 μM cyclosporine A (a general transporter inhibitor47), we observed that Fic of simvastatin acid was constant across all concentrations and lower than previously observed without inhibition (Fig. 3c). This suggests that additional active transport mechanisms (including uptake transporters) are present in these cells and affect the cellular disposition of this compound.
To better understand these observations, we simulated cellular drug accumulation using three simple kinetic models: 1) one including both active uptake and active efflux mechanisms (corresponding to P-gp transfected cells), 2) one including only the uptake mechanisms (corresponding to cP-gp-KO cells), and 3) one including only the passive mechanism (corresponding to inhibition of all active mechanisms with cyclosporine A). We simulated all possible combinations of low, medium and high values of passive permeability (0.1, 1, and 10 × 10−6 cm/s), Vmax (10, 100, and 1000 pmol/min/mg protein) and Km (1, 10, and 100 μM) for each of the transporters (see Matsson et al.48 and the ‘Methods’ section for details on the simulations). The simulations were in best agreement with the experimental observations for a compound with high passive permeability (10 × 10−6 cm/s), medium affinity (Km = 10 μM) for an uptake transporter and lower affinity (Km = 100 μM) for an efflux transporter, with both transporters having comparable (high) transport capacities (Vmax) (Fig. 3d). This matches previous observations for simvastatin acid, which show that this compound, in addition to being an OATP1B1 substrate, has a high passive permeability, and a low affinity for efflux transporters49,50,51.
Intracellular bioavailability (Fic) in complex cell systems
Our results in MDCK cells revealed that unknown endogenous transport mechanisms can influence Fic to a larger extent than previously recognized. To further investigate how multiple transporters impact Fic, we performed measurements in freshly isolated human hepatocytes. These cells also express metabolizing enzymes, which can further affect Fic (Fig. 1), and better represent an in vivo situation where multiple processes occur simultaneously. Hepatocytes were used in two commonly employed culture conditions (Fig. 4a): 1) freshly isolated cells (in suspension) that express similar levels of transporters and metabolizing enzymes as those observed in the human liver29,30,31,52; and 2) cells cultured for 24 h post-isolation (in monolayer format) with a significant down-regulation of many important transporters and enzymes due to cell dedifferentiation29,30,31. Thus, without knowing the exact composition of the contributing transporters and enzymes, their relative impact in two complex hepatocyte systems could be compared. For these studies, we selected a subset of 16 compounds from the initial dataset (Supplementary Table 4). The compounds were chosen to be structurally diverse and to be substrates of different transporters and enzymes with altered expression (Supplementary Table 5).

(a) Schematic representation of hepatocyte cultures used in this study: freshly isolated human hepatocytes in suspension show higher activity of drug-transporting proteins and metabolizing enzymes than hepatocytes cultured for 24 h in monolayer format. (b) Comparison between Fic in suspension hepatocytes and hepatocytes cultured in monolayer format. Negatively charged compounds at pH 7.4 are represented as triangles, neutral and zwitterionic species are represented by circles, and positively charged compounds are represented by squares. Compound numbers indicate: 1) astemizole; 2) indomethacin; 3) ketoconazole; 4) atorvastatin acid; 5) propranolol; 6) ritonavir. (c) Differences in metabolic clearance in suspension hepatocytes and hepatocytes cultured in monolayer format. *p < 0.05 in Mann-Whitney test comparing suspension and monolayer hepatocytes.
On average, Fic was 5.7-fold lower in freshly isolated suspension hepatocytes than in monolayer hepatocytes cultivated for 24 h (Fig. 4b; p = 0.0002 in Wilcoxon matched-pairs signed rank test). No apparent correlation was observed between the physicochemical characteristics of the compounds and the differences in Fic in the two types of hepatocytes (Supplementary Figure 5). However, compounds that are reported as substrates of multiple efflux transporters and enzymes displayed larger differences between the two culturing conditions (rS = 0.72 for the rank-order correlation between differences in Fic and the number of targeted enzymes and transporters). This was in agreement with the reported higher mRNA transcript levels in suspension hepatocytes for these proteins29,30,31, which are associated with the removal of compounds from the cell interior (leading to a lower Fic). In agreement with these results, compounds that were metabolically cleared at higher rates (e.g., atorvastatin, propranolol, and ritonavir; Fig. 4c; p < 0.05 in Mann-Whitney test) showed lower Fic in suspension hepatocytes. In analogy, compounds with a similar metabolic clearance under both culture conditions (e.g., astemizole, indomethacin, and ketoconazole) also had similar Fic (Fig. 4c). These results strongly support our hypothesis that Fic is a general predictor of intracellular drug disposition. We also measured the protein concentrations of the major transporters and metabolic enzymes involved in the clearance of the studied compounds using state-of-the-art mass spectrometry-based proteomics (Supplementary Figure 6). To our surprise, the agreement between mRNA transcript levels29,30, enzymatic activity (Fig. 4c) and Fic (Fig. 4b) did not translate to the protein level, since we did not observe significant differences in protein concentrations between the two culturing conditions (p = 0.6 in Wilcoxon matched-pairs signed rank test). This can be explained by drug-transporting proteins being redistributed from the plasma membrane to intracellular compartments under certain culturing conditions, or as a result of external stimuli53,54,55. Similarly, the activity of metabolic enzymes can be reduced by post-translational modifications56 or lower levels of co-factors57. In these cases, these proteins will contribute to the whole-cell protein levels but will not affect Fic. This underscores the importance of functional studies to assess the impact of drug-transporting proteins and metabolizing enzymes on intracellular drug concentrations.
Discussion
Intracellular compound exposure is of paramount importance for the pharmacodynamics of drugs acting on intracellular targets, and for interactions with intracellular drug-metabolizing enzymes11,28. In this study, we introduce the concept of intracellular compound bioavailability (Fic) and demonstrate its utility in describing the impact of drug-transporting proteins and metabolizing enzymes on cellular drug disposition.
Our methodology is based on the parallel determination of the fraction of intracellular compound that is not bound to cellular components (fu,cell) and cellular compound accumulation (Kp). Our results indicate that overexpression of single proteins (e.g. OATP1B1) has little impact on fu,cell (Fig. 2b). Further, good correlations were observed for fu,cell across all cell types used in this study, despite different proteomes in these cells (rS > 0.8 for all comparisons of between HEK293 cells, MDCK cells and freshly isolated hepatocytes). This supports previous observations that fu,cell is mostly dominated by membrane partitioning25,26,32,33,34, and that specific proteins contribute negligibly to this parameter.
In contrast, clear effects were observed for Fic when HEK293 cells were transfected with the cellular uptake-mediating transporter OATP1B1, with intracellular exposure increasing for all investigated substrates of OATP1B1. Interestingly, some statins, such as atorvastatin and pitavastatin, showed Fic < 0.1 when the transporter was lacking, suggesting that they have limited cell penetration in the absence of transporters.
Our studies on the concentration-dependent uptake of the OATP1B1 substrates showed a greater impact of the transporter at lower substrate concentrations. At the lowest measured concentrations (approaching unbound clinical plasma concentrations), Fic was up to 10-fold higher for atorvastatin and pitavastatin, and up to 2-fold higher for fluvastatin and simvastatin acid in the presence of the transporter. These results are in agreement with clinical drug-drug interaction studies12,13,37,38, in which the pharmacokinetics of atorvastatin and pitavastatin are more affected than fluvastatin and simvastatin acid when co-administered with OATP1B1 inhibitors. For example, cyclosporine A increases the AUC of pitavastatin and atorvastatin by 5- to 15- fold, and that of simvastatin and fluvastatin by 3- to 4-fold in human subjects58. Furthermore, pharmacogenomic studies have also shown that reduced-function haplotypes of SLCO1B1 contribute to higher plasma concentrations of these compounds59,60,61. With the exception of simvastatin acid, which (based on only one study62) has a larger clinical effect than the other statins from a reduced function polymorphism of OATP1B1, our results accurately reproduce the rank order of the clinically observed AUC change (pitavastatin > atorvastatin > fluvastatin).
Contrary to HEK293 cells, MDCK cells express significant levels of endogenous drug-transporting proteins, including canine P-gp, that complicate the interpretation of in vitro transport studies42,63. To abolish the impact of canine P-gp in the present study, we used our recently established MDCK cell line where this transporter was completely knocked out using CRISPR-Cas9 (cP-gp-KO cells)44. This enabled the study of baseline transport into MDCK cells without the need for chemical inhibition of P-gp. This was considered important, since we have shown that P-gp inhibitors such as elacridar also inhibit other endogenous transporters35,47. Since efflux transporters are driven by intracellular (or intramembraneous) substrate concentrations, it is desirable to avoid simultaneous inhibition of the uptake transporters that some compounds depend on to reach the cell interior. Thus, studies that rely on chemical inhibition can misclassify compounds as non-substrates. In fact, we observed that some compounds not previously annotated as P-gp substrates (e.g. astemizole, ketoconazole, lovastatin and rosiglitazone) showed large differences in Fic between P-gp-expressing and cP-gp-KO cell lines. These compounds have previously been reported as P-gp inhibitors64,65,66. Our findings suggest that they are also transported substrates. In follow-up experiments, we have confirmed that at least two of these compounds show asymmetrical transport across a monolayer of MDCK cells (manuscript in preparation). This indicates that the setup used here can replace more demanding transport assays to identify substrates of proteins involved in drug disposition and resistance to chemotherapy67.
The possibility of studying the involvement of multiple transport processes with this methodology, as observed with simvastatin acid in MDCK cells (Fig. 3c), prompted us to study Fic in the more complex system of freshly isolated human hepatocytes. Primary human hepatocyte models are extensively used in drug discovery and are available in many different configurations, which display marked differences in gene expression and activity of drug-transporting proteins and drug-metabolizing enzymes29,30,31. Our measurements of Fic reflected these differences in that lower Fic values were observed in suspension hepatocytes, where the elimination pathways via efflux transporters and drug-metabolizing enzymes are more active29,30,31. Interestingly, only compounds reported to be substrates of multiple drug-transporting proteins and metabolizing enzymes were affected (Supplementary Table 5), while compounds reported to be substrates of mainly one or two metabolizing enzymes (e.g. astemizole, indinavir, ketoconazole, lopinavir, and rosiglitazone) displayed similar Fic in both culturing conditions. For example, ritonavir, a compound with an Fic of 1.0 in mock-transfected HEK293 cells (where passive mechanisms are the major determinants of drug accumulation25), showed an Fic of 0.26 in freshly isolated suspension hepatocytes (indicative of a dominance of elimination mechanisms) and an Fic of 2.3 in monolayer hepatocytes (indicative of reduced elimination and a dominance of active uptake mechanisms). This was confirmed by functional in vitro clearance studies, which showed that the metabolic CLint of ritonavir was in fact reduced in monolayer hepatocytes (Fig. 4c). Thus, our results indicate that Fic can be used to assess intracellular drug exposure also in complex systems with multiple sequential and parallel drug disposition processes.
Previous approaches to determine intracellular unbound drug concentrations based on mathematical modeling require extensive collection of experimental data (at multiple time points and concentrations)3,4,21, which limits their compound throughput. Alternative approaches rely on a separate measurement of drug binding to the cells that is combined measurements of the total intracellular drug concentrations. In such approaches, binding can be estimated by inhibiting all active processes (with chemical inhibitors24,68 or reduced temperature21) or using equilibrium dialysis of tissue or cell homogenates22,25. The use of chemical inhibitors assumes that all transport and metabolism mechanisms are known and can be inhibited, while lowering the temperature assumes that changes in membrane fluidity69 do not affect drug binding. Our approach uses equilibrium dialysis of cell homogenates and assumes that binding is not affected by the disruption of the cellular context during homogenization. This allows the processing of large numbers of compounds without prior knowledge of compound elimination kinetics or metabolic pathways. Despite the utility of a simple estimate of intracellular available compound, the Fic parameter does not provide information on the subcellular distribution of the compound. Instead, it provides an average bioavailability in the whole cell. As previously shown, mathematical modeling of pH22,25 or electrochemical gradients5 can offer insight into the accumulation of compounds in lysosomes or mitochondria. In addition, inhibition of ion channels responsible for the maintenance of these gradients can be used to validate such models25. Alternatively, subcellular fractionation can be performed prior to70 or after20 incubation with the drug. However, fractionation approaches might alter compound distribution, since the organelle is no longer in its native environment. All of these approaches are compatible with the overall methodology presented in this study and can be used in specific cases, when the target of interest is located in a particular subcellular compartment.
In conclusion, we introduce Fic as a new approach to study the impact of proteins involved in drug disposition on the intracellular drug concentration available for target and off-target (e.g. drug-metabolizing enzymes) interactions. We show how single and multiple drug-transporting proteins and metabolizing enzymes influence Fic in various cellular systems of different complexity. We also show that our setup enables identification of potential substrates of transporters and could constitute an alternative to state-of-the-art in vitro permeability assays. Thus, Fic provides a true value of intracellular accumulation that is unbiased from membrane interactions. Fic should therefore not only be applicable in predictive pharmacokinetics, but in particular during compound profiling in drug discovery aiming at intracellular targets11,27,28,71,72.
Methods
Theoretical background: intracellular compound bioavailability (Fic)
To determine intracellular unbound compound concentrations, we measured intracellular fraction of unbound compound (fu,cell) in parallel with total intracellular compound accumulation (Ccell), as described below. Intracellular compound bioavailability (Fic) was calculated as the ratio between intracellular unbound compound concentration (Cu,cell = fu,cell∙Ccell) (the concentration available to bind intracellular targets) and the externally added compound concentration (Cmedium):

where Kp is the ratio Ccell/Cmedium.
In our previous work25, this ratio was termed unbound drug accumulation ratio (Kpuu), in analogy to previous work on unbound drug exposure in brain tissue73. We introduce the more descriptive and general Fic parameter here, since it is applicable also to situations where only a fraction of the extracellular compound is unbound. This is of interest in drug discovery settings where cellular screens are often performed in the presence of serum proteins. In such assay setups, the Kpuu term is not applicable, since it assumes extracellular drug to be completely unbound.
Contrary to oral bioavailability, which is defined in terms of dose (i.e., amount), Fic is defined in concentration terms. For that reason, Fic can take any positive value and is not limited to the range between zero and one. However, Fic can be interpreted in similar terms, as the fraction of the extracellular concentration that is available intracellularly.
Materials and compound set
Cell culture reagents were acquired from Thermo Fisher Scientific Inc. or Sigma-Aldrich. HEK293 cells were originally obtained from Thermo Fisher Scientific and MDCK cells from ATCC. Test compounds of analytical grade (≥95% purity) were obtained from Sigma-Aldrich, Toronto Research Chemicals or OChem Inc. Compounds were dissolved in dimethyl sulfoxide (DMSO) and stored at −20 °C.
The compound set used in this study included 34 drug-like molecules (Supplementary Table 1), and was enriched in compounds previously described as OATP1B1 (n = 11)74 or P-gp substrates (n = 22)75,76, with ten of the compounds included being substrates of both drug-transporting proteins. ADMET Predictor, version 7.0 (SimulationsPlus, Lancaster, CA) was used to predict physicochemical characteristics of the compound set.
The compounds were generally within the boundaries of Lipinski’s rule of 5 (64% of molecules within all rule-of-5 boundaries77), with the major violation being MW > 500 Da (83% of violations). At physiological pH (pH = 7.4), eight of the included compounds were predicted to be predominantly negatively charged, fourteen as mostly uncharged, nine as mostly positively charged, and three as zwitterions (Supplementary Table 1). In agreement with the literature74, substrates of OATP1B1 were generally negatively charged and more polar than non-substrates (mean PSA of 105 vs. 74 Å2; p = 0.03). Compounds previously described as substrates of P-gp were more polar (mean PSA of 99 vs. 56 Å2; p = 0.002) and larger (mean MW of 497 vs. 335; p < 0.001) than compounds lacking reports of P-gp-mediated transport.
Cell culture and preparation of experiments
Cell line maintenance
HEK293 cells stably transfected with OATP1B1 (SLCO1B1*1a allele) or with the corresponding empty vector were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine and 75 μg/ml hygromycin B.78 CRISPR-Cas9 canine P-gp knock-out MDCK cells (cP-gp-KO) were generated as described elsewhere44. MDCK cells were grown in DMEM (glucose 1 g/l) supplemented with 10% FBS, GlutaMAX, penicillin (100 units/ml), and streptomycin (100 μg/ml). For human P-gp (ABCB1) transfected MDCK cells, hygromycin B (375 μg/ml) was included as a selection antibiotic. All cell lines were kept at 37 °C in a humidified 5% CO2 atmosphere and subcultivated twice a week with a 1:6 ratio.
Isolation of primary human hepatocytes
Hepatocytes were isolated from human liver tissue obtained from two donors undergoing surgical resections at the Department of Surgery, Uppsala University Hospital. Ethical approval was granted by the Uppsala Regional Ethics Committee (ethical approvals no. 2009/028 and 2011/037), donors gave informed consent and all studies were performed in accordance with the current national regulations and ethical guidelines. Isolation was performed using a two-step collagenase procedure, as previously described52,79.
Preparation of cell homogenates for cell binding experiments
For intracellular fraction of unbound compound experiments, cell lines or primary cells were suspended to a concentration of 10 × 106 cells/ml in Hank’s balanced salt solution (HBSS). Cell suspensions were homogenized for 10 s with a VCX-500 ultrasonic processor (Sonics & Materials) at 20% intensity and used immediately for binding experiments25.
Preparation for intracellular compound accumulation experiments
HEK293 cells were grown for two days in 24-well plates, as previously described25. MDCK cells were grown for two days in 96-well plates, prior to the intracellular compound accumulation experiments. The growth media were the same as described in the section ‘Cell line maintenance’ without addition of hygromycin B to avoid transporter inhibition.
For the corresponding experiments in human hepatocytes, two cell culture configurations were used: freshly isolated cells in suspension, or cells cultivated as monolayers for 24 h. The freshly isolated cells were suspended directly after isolation at a concentration of 5 × 106 cells/ml in HBSS. The cell suspension (100 μl) was added to 96-well plates, which were immediately used for accumulation experiments. The monolayer-cultured hepatocytes were allowed to attach to collagen-coated 24-well plates for three hours (375 000 cells/well) in DMEM with HEPES supplemented with 5% FBS, penicillin (100 units/ml), streptomycin (100 μg/ml), insulin (4 μg/ml), and dexamethasone (1 μM). After cell attachment, medium was replaced by hepatocyte maintenance medium (Lonza) supplemented with insulin (10 μg/ml), transferrin (5.5 μg/ml), selenium (5 ng/ml), penicillin (100 units/ml), streptomycin (100 μg/ml), and dexamethasone (0.1 μM). Cells were kept in maintenance medium for 24 h, until the start of the intracellular compound accumulation experiments.
Intracellular fraction of unbound compound
Binding to cell homogenates at steady-state was measured using dialysis as previously described26. Simultaneous measurement of intracellular binding for multiple compounds (cassette-mode) is made possible by our previous observation that binding to cellular structures at low compound concentration is mainly non-specific25 and unaffected by the presence of other drug-like molecules26. Briefly, the cell homogenate was spiked with six randomly-chosen compounds. Homogenates were dialyzed for 4 h using a Rapid Equilibrium Dialysis device (Thermo Fisher Scientific Inc.). At the end of the incubation, uniform sample matrices were obtained by addition of blank homogenate to samples from the buffer chamber, and by addition blank buffer to samples from the homogenate chamber. Dilutions of samples from the homogenate chambers (10-fold and 100-fold) were prepared. Protein was precipitated with acetonitrile/water (60:40) spiked with 50 nM warfarin (internal standard). Samples were centrifuged for 20 min at 2465 × g. Compounds in the supernatant were analyzed by ultra-performance liquid chromatography coupled to tandem mass spectrometry (UPLC-MS/MS), as described below.
The fraction of compound that was not bound to cell homogenate (fu,hom) was calculated according to:

where PAbuffer is the peak area of compound in the buffer chamber and PAhom is the peak area of compound in the homogenate chamber, both corrected for the peak area of the internal standard in the respective chamber. For each compound, the dilution of the homogenate sample where PAhom was closest to PAbuffer was used.
The fraction of unbound compound in the cell (fu,cell) was calculated according to:

where D was estimated for each homogenate preparation based on a cellular volume of 6.5 μl/mg protein80, and on the protein concentration measured using the BCA Protein Assay Reagent Kit (Thermo Fisher Scientific Inc.).
Intracellular compound accumulation
Intracellular compound accumulation at steady-state was measured essentially as described before25. Briefly, cells were washed twice with HBSS and incubated for 45 min with solutions of single test compounds (in HBSS) on an orbital shaker (300 rpm) at 37 °C. At the end of the experiment, the remaining extracellular compound solutions were collected and cells were washed twice with ice-cold phosphate-buffered saline pH 7.4 (PBS). For hepatocytes in suspension, the cells were separated from the extracellular solutions by centrifugation (100 × g for 5 min). Compounds were extracted from the cells using acetonitrile/water (60:40) spiked with 50 nM warfarin (internal standard). Extracellular medium samples were diluted 10-fold with the same extraction solution. All samples were centrifuged for 20 min at 2465 × g and compounds were quantified in the supernatant by UPLC-MS/MS, as described below. Protein concentrations were measured in representative wells of the plates using the BCA Protein Assay Reagent Kit (Thermo Fisher Scientific Inc.).
The ratio between total compound concentrations in the cells and in the medium (Kp) was calculated according to:

where Acell is the amount of compound in the cell samples, Vcell is the cell volume (6.5 μL/mg protein80), and Cmedium is the compound concentration in the medium sample.
Metabolic clearance (CLint) in primary human hepatocytes
Metabolic clearance (CLint) was determined, as previously described81. Briefly, freshly isolated human hepatocytes (in suspension or after 24 h of culture in monolayer format) were incubated with 1 μM compound solution (total incubation volume = 200 μl). At time points 0, 10, 30, and 60 min, the reaction was stopped by addition of acetonitrile (200 μl) spiked with 50 nM warfarin (internal standard). Samples were then centrifuged for 20 min at 2465 × g and compounds were quantified in the supernatant by UPLC-MS/MS, as described below. CLint was calculated as the product of the compound disappearance rate and incubation volume, normalized by the cell number.
Compound quantification
Compound quantification was performed using UPLC-MS/MS. The system consisted of a Waters Xevo TQ MS with electrospray ionization coupled to a Waters Acquity UPLC. Compounds were chromatographically separated with a 2 min gradient elution (flow rate of 0.5 ml/min) on a Waters BEH C18 column, 2.1 × 50 mm (1.7 μm) at 60 °C, as described elsewhere25. Mass transitions for each compound and their respective cone voltages and collision energies can be found in Supplementary Table 7.
Protein quantification of drug-transporting proteins and drug-metabolizing enzymes
Protein concentrations for drug-transporting proteins and drug-metabolizing enzymes were quantified as previously described44. Briefly, a cell pellet (10 × 106 cells) of freshly isolated human hepatocytes or of cells cultured in monolayer format was lysed with 100 mM Tris/HCl, pH 7.8, 2% (m/v) SDS and 50 mM DTT. Proteins were denatured at 95 °C for 5 min. The lysates were sonicated and then clarified by centrifugation. Trypsin digestion was performed using the filter-aided sample preparation protocol82. Peptides were eluted and spiked with stable-isotope labelled standard peptides (JPT Peptide Technologies) and analyzed with UPLC-MS/MS as previously described44. The sequences of proteospecific peptides have been previously published83,84.
Intracellular compound accumulation simulations
Intracellular compound accumulation was simulated, as previously described48, with a mechanistic model developed in R version 3.1.1 (http://www.r-project.org) using the deSolve package for differential equation solving. Integration was performed using the ode function, which automatically selects an integration algorithm of appropriate stiffness. The model consisted of two compartments (extracellular and intracellular), with model dimensions set to reflect the setup used in accumulation experiments in MDCK cells (apical surface area = 1 × 10−4 m2, extracellular volume = 200 μl, total intracellular volume = 0.25 μl). In simulations intended to reflect compound accumulation in MDCK P-gp knockout cells, compound transport across the apical membrane was incorporated as a combination of transporter-mediated uptake (assumed to follow regular Michaelis-Menten kinetics) and bidirectional passive Fick’s diffusion. Simulations of compound accumulation in MDCK cells transfected with human P-gp also included a saturable efflux component in the apical membrane, while simulations of compound accumulation in cells where active transport has been inhibited with cyclosporine A included only bidirectional passive Fick’s diffusion. All combinations of low, medium, and high values of passive permeability (0.1, 1, and 10 × 10−6 cm/s), and Vmax (10, 100, and 1000 pmol/min/mg protein) and Km (1, 10, and 100 μM) for each of the transporters were simulated. Each simulation was repeated at 20 concentrations in the range 0.01 μM–100 μM. Drug accumulation was simulated over 120 min, and the resulting equilibrium intra- and extracellular unbound concentrations were used to calculate Fic. The concentration dependencies of Fic were then visually inspected to identify scenarios that matched experimental observations.
Statistics
All experiments were performed in triplicate on at least 2 independent occasions. Atorvastatin and lopinavir were used as controls in intracellular fraction of unbound compound experiments (added to all cassettes: average coefficient of variation (CV) in a single experiment <20%; day-to-day CV < 10%), and in intracellular compound accumulation experiments (in parallel with tested compound: average CV in a single experiment <10%; day-to-day CV < 10%). All results are presented as geometric mean ± standard error of the mean (S.E.M.), unless otherwise stated. A p-value < 0.05 was considered significant.
Additional Information
How to cite this article: Mateus, A. et al. Intracellular drug bioavailability: a new predictor of system dependent drug disposition. Sci. Rep. 7, 43047; doi: 10.1038/srep43047 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
International Transporter Consortium et al. Membrane transporters in drug development. Nat Rev Drug Discov 9, 215–236 (2010).
Chu, X. et al. Intracellular drug concentrations and transporters: measurement, modeling, and implications for the liver. Clin Pharmacol Ther 94, 126–141 (2013).
Yabe, Y., Galetin, A. & Houston, J. B. Kinetic characterization of rat hepatic uptake of 16 actively transported drugs. Drug Metab Dispos 39, 1808–1814 (2011).
Menochet, K., Kenworthy, K. E., Houston, J. B. & Galetin, A. Simultaneous assessment of uptake and metabolism in rat hepatocytes: a comprehensive mechanistic model. J Pharmacol Exp Ther 341, 2–15 (2012).
Chien, H. C. et al. Rapid Method To Determine Intracellular Drug Concentrations in Cellular Uptake Assays: Application to Metformin in Organic Cation Transporter 1-Transfected Human Embryonic Kidney 293 Cells. Drug Metab Dispos 44, 356–364 (2016).
Nakagawa, M. et al. Reduced intracellular drug accumulation in the absence of P-glycoprotein (mdr1) overexpression in mitoxantrone-resistant human MCF-7 breast cancer cells. Cancer Res 52, 6175–6181 (1992).
Widmer, N., Colombo, S., Buclin, T. & Decosterd, L. A. Functional consequence of MDR1 expression on imatinib intracellular concentrations. Blood 102, 1142 (2003).
Korzekwa, K. R., Nagar, S., Tucker, J., Weiskircher, E. A., Bhoopathy, S. & Hidalgo, I. J. Models to Predict Unbound Intracellular Drug Concentrations in the Presence of Transporters. Drug Metab Dispos 40, 865–876 (2012).
Pang, K. S., Maeng, H. J. & Fan, J. Interplay of transporters and enzymes in drug and metabolite processing. Mol Pharm 6, 1734–1755 (2009).
Brown, H. S., Wilby, A. J., Alder, J. & Houston, J. B. Comparative use of isolated hepatocytes and hepatic microsomes for cytochrome P450 inhibition studies: transporter-enzyme interplay. Drug Metab Dispos 38, 2139–2146 (2010).
Morgan, P. et al. Can the flow of medicines be improved? Fundamental pharmacokinetic and pharmacological principles toward improving Phase II survival. Drug Discov Today 17, 419–424 (2012).
Vildhede, A. et al. Hepatic uptake of atorvastatin: influence of variability in transporter expression on uptake clearance and drug-drug interactions. Drug Metab Dispos 42, 1210–1218 (2014).
Vildhede, A. et al. Mechanistic Modeling of Pitavastatin Disposition in Sandwich-Cultured Human Hepatocytes: A Proteomics-Informed Bottom-Up Approach. Drug Metab Dispos 44, 505–516 (2016).
Ikema, S. et al. Okadaic acid is taken-up into the cells mediated by human hepatocytes transporter OATP1B3. Food Chem Toxicol 83, 229–236 (2015).
Chen, Y. et al. Effect of genetic variation in the organic cation transporter 2 on the renal elimination of metformin. Pharmacogenet Genomics 19, 497–504 (2009).
Liu, X. et al. Biliary excretion in primary rat hepatocytes cultured in a collagen-sandwich configuration. Am J Physiol 277, G12–21 (1999).
Hewitt, N. J. et al. Primary hepatocytes: current understanding of the regulation of metabolic enzymes and transporter proteins, and pharmaceutical practice for the use of hepatocytes in metabolism, enzyme induction, transporter, clearance, and hepatotoxicity studies. Drug Metab Rev 39, 159–234 (2007).
Olinga, P. et al. The applicability of rat and human liver slices to the study of mechanisms of hepatic drug uptake. J Pharmacol Toxicol Methods 45, 55–63 (2001).
Li, N. et al. Development of an improved three-dimensional in vitro intestinal mucosa model for drug absorption evaluation. Tissue Eng Part C Methods 19, 708–719 (2013).
Pfeifer, N. D., Harris, K. B., Yan, G. Z. & Brouwer, K. L. Determination of intracellular unbound concentrations and subcellular localization of drugs in rat sandwich-cultured hepatocytes compared with liver tissue. Drug Metab Dispos 41, 1949–1956 (2013).
Nordell, P., Winiwarter, S. & Hilgendorf, C. Resolving the distribution-metabolism interplay of eight OATP substrates in the standard clearance assay with suspended human cryopreserved hepatocytes. Mol Pharm 10, 4443–4451 (2013).
Friden, M. et al. Measurement of unbound drug exposure in brain: modeling of pH partitioning explains diverging results between the brain slice and brain homogenate methods. Drug Metab Dispos 39, 353–362 (2011).
Loryan, I., Friden, M. & Hammarlund-Udenaes, M. The brain slice method for studying drug distribution in the CNS. Fluids Barriers CNS 10, 6 (2013).
Keemink, J., Augustijns, P. & Annaert, P. Unbound ritonavir concentrations in rat and human hepatocytes. J Pharm Sci 104, 2378–2387 (2015).
Mateus, A., Matsson, P. & Artursson, P. Rapid measurement of intracellular unbound drug concentrations. Mol Pharm 10, 2467–2478 (2013).
Mateus, A., Matsson, P. & Artursson, P. A high-throughput cell-based method to predict the unbound drug fraction in the brain. J Med Chem 57, 3005–3010 (2014).
Gordon, L. J. et al. Direct Measurement of Intracellular Compound Concentration by RapidFire Mass Spectrometry Offers Insights into Cell Permeability. J Biomol Screen 21, 156–164 (2016).
Almqvist, H. et al. CETSA screening identifies known and novel thymidylate synthase inhibitors and slow intracellular activation of 5-fluorouracil. Nat Commun 7, 11040 (2016).
LeCluyse, E. L. Human hepatocyte culture systems for the in vitro evaluation of cytochrome P450 expression and regulation. Eur J Pharm Sci 13, 343–368 (2001).
Richert, L. et al. Gene expression in human hepatocytes in suspension after isolation is similar to the liver of origin, is not affected by hepatocyte cold storage and cryopreservation, but is strongly changed after hepatocyte plating. Drug Metab Dispos 34, 870–879 (2006).
Lundquist, P. et al. Functional ATP-binding cassette drug efflux transporters in isolated human and rat hepatocytes significantly affect assessment of drug disposition. Drug Metab Dispos 42, 448–458 (2014).
Bickel, M. H. & Steele, J. W. Binding of basic and acidic drugs to rat tissue subcellular fractions. Chem Biol Interact 8, 151–162 (1974).
Longhi, R. et al. Brain tissue binding of drugs: evaluation and validation of solid supported porcine brain membrane vesicles (TRANSIL) as a novel high-throughput method. Drug Metab Dispos 39, 312–321 (2011).
Nagar, S. & Korzekwa, K. Commentary: nonspecific protein binding versus membrane partitioning: it is not just semantics. Drug Metab Dispos 40, 1649–1652 (2012).
Karlgren, M. et al. Classification of inhibitors of hepatic organic anion transporting polypeptides (OATPs): influence of protein expression on drug-drug interactions. J Med Chem 55, 4740–4763 (2012).
Harwood, M. D., Russell, M. R., Neuhoff, S., Warhurst, G. & Rostami-Hodjegan, A. Lost in centrifugation: accounting for transporter protein losses in quantitative targeted absolute proteomics. Drug Metab Dispos 42, 1766–1772 (2014).
Watanabe, T. et al. Investigation of the rate-determining process in the hepatic elimination of HMG-CoA reductase inhibitors in rats and humans. Drug Metab Dispos 38, 215–222 (2010).
Shitara, Y., Maeda, K., Ikejiri, K., Yoshida, K., Horie, T. & Sugiyama, Y. Clinical significance of organic anion transporting polypeptides (OATPs) in drug disposition: their roles in hepatic clearance and intestinal absorption. Biopharm Drug Dispos 34, 45–78 (2013).
Camenisch, G., Alsenz, J., van de Waterbeemd, H. & Folkers, G. Estimation of permeability by passive diffusion through Caco-2 cell monolayers using the drugs’ lipophilicity and molecular weight. Eur J Pharm Sci 6, 317–324 (1998).
Linnankoski, J., Ranta, V. P., Yliperttula, M. & Urtti, A. Passive oral drug absorption can be predicted more reliably by experimental than computational models—fact or myth. Eur J Pharm Sci 34, 129–139 (2008).
Sugano, K. et al. Coexistence of passive and carrier-mediated processes in drug transport. Nat Rev Drug Discov 9, 597–614 (2010).
Ahlin, G., Hilgendorf, C., Karlsson, J., Szigyarto, C. A., Uhlen, M. & Artursson, P. Endogenous gene and protein expression of drug-transporting proteins in cell lines routinely used in drug discovery programs. Drug Metab Dispos 37, 2275–2283 (2009).
Sharma, P., Butters, C. J., Smith, V., Elsby, R. & Surry, D. Prediction of the in vivo OATP1B1-mediated drug-drug interaction potential of an investigational drug against a range of statins. Eur J Pharm Sci 47, 244–255 (2012).
Simoff, I. et al. Complete Knockout of Endogenous Mdr1 (Abcb1) in MDCK Cells by CRISPR-Cas9. J Pharm Sci 105, 1017–1021 (2016).
Gatlik-Landwojtowicz, E., Aanismaa, P. & Seelig, A. Quantification and characterization of P-glycoprotein-substrate interactions. Biochemistry 45, 3020–3032 (2006).
Aller, S. G. et al. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 323, 1718–1722 (2009).
Matsson, P., Pedersen, J. M., Norinder, U., Bergstrom, C. A. & Artursson, P. Identification of novel specific and general inhibitors of the three major human ATP-binding cassette transporters P-gp, BCRP and MRP2 among registered drugs. Pharm Res 26, 1816–1831 (2009).
Matsson, P., Fenu, L. A., Lundquist, P., Wisniewski, J. R., Kansy, M. & Artursson, P. Quantifying the impact of transporters on cellular drug permeability. Trends Pharmacol Sci 36, 255–262 (2015).
Hochman, J. H. et al. Interactions of human P-glycoprotein with simvastatin, simvastatin acid, and atorvastatin. Pharm Res 21, 1686–1691 (2004).
Chen, C. et al. Differential interaction of 3-hydroxy-3-methylglutaryl-coa reductase inhibitors with ABCB1, ABCC2, and OATP1B1. Drug Metab Dispos 33, 537–546 (2005).
Neve, E. P., Artursson, P., Ingelman-Sundberg, M. & Karlgren, M. An integrated in vitro model for simultaneous assessment of drug uptake, metabolism, and efflux. Mol Pharm 10, 3152–3163 (2013).
Vildhede, A., Wisniewski, J. R., Noren, A., Karlgren, M. & Artursson, P. Comparative Proteomic Analysis of Human Liver Tissue and Isolated Hepatocytes with a Focus on Proteins Determining Drug Exposure. J Proteome Res 14, 3305–3314 (2015).
Hoffmaster, K. A., Turncliff, R. Z., LeCluyse, E. L., Kim, R. B., Meier, P. J. & Brouwer, K. L. P-glycoprotein expression, localization, and function in sandwich-cultured primary rat and human hepatocytes: relevance to the hepatobiliary disposition of a model opioid peptide. Pharm Res 21, 1294–1302 (2004).
Kim, H., Barroso, M., Samanta, R., Greenberger, L. & Sztul, E. Experimentally induced changes in the endocytic traffic of P-glycoprotein alter drug resistance of cancer cells. Am J Physiol 273, C687–702 (1997).
Porcelli, L., Lemos, C., Peters, G. J., Paradiso, A. & Azzariti, A. Intracellular trafficking of MDR transporters and relevance of SNPs. Curr Top Med Chem 9, 197–208 (2009).
Aguiar, M., Masse, R. & Gibbs, B. F. Regulation of cytochrome P450 by posttranslational modification. Drug Metab Rev 37, 379–404 (2005).
Yamazaki, H., Shimada, T., Martin, M. V. & Guengerich, F. P. Stimulation of cytochrome P450 reactions by apo-cytochrome b5: evidence against transfer of heme from cytochrome P450 3A4 to apo-cytochrome b5 or heme oxygenase. J Biol Chem 276, 30885–30891 (2001).
Kalliokoski, A. & Niemi, M. Impact of OATP transporters on pharmacokinetics. Br J Pharmacol 158, 693–705 (2009).
Niemi, M. Transporter pharmacogenetics and statin toxicity. Clin Pharmacol Ther 87, 130–133 (2010).
Wilke, R. A. et al. The clinical pharmacogenomics implementation consortium: CPIC guideline for SLCO1B1 and simvastatin-induced myopathy. Clin Pharmacol Ther 92, 112–117 (2012).
Maeda, K. Organic anion transporting polypeptide (OATP)1B1 and OATP1B3 as important regulators of the pharmacokinetics of substrate drugs. Biol Pharm Bull 38, 155–168 (2015).
Pasanen, M. K., Neuvonen, M., Neuvonen, P. J. & Niemi, M. SLCO1B1 polymorphism markedly affects the pharmacokinetics of simvastatin acid. Pharmacogenet Genomics 16, 873–879 (2006).
Kuteykin-Teplyakov, K., Luna-Tortos, C., Ambroziak, K. & Loscher, W. Differences in the expression of endogenous efflux transporters in MDR1-transfected versus wildtype cell lines affect P-glycoprotein mediated drug transport. Br J Pharmacol 160, 1453–1463 (2010).
Weiss, J., Sauer, A., Herzog, M., Boger, R. H., Haefeli, W. E. & Benndorf, R. A. Interaction of thiazolidinediones (glitazones) with the ATP-binding cassette transporters P-glycoprotein and breast cancer resistance protein. Pharmacology 84, 264–270 (2009).
Keogh, J. P. & Kunta, J. R. Development, validation and utility of an in vitro technique for assessment of potential clinical drug-drug interactions involving P-glycoprotein. Eur J Pharm Sci 27, 543–554 (2006).
Bogman, K., Peyer, A. K., Torok, M., Kusters, E. & Drewe, J. HMG-CoA reductase inhibitors and P-glycoprotein modulation. Br J Pharmacol 132, 1183–1192 (2001).
Ambudkar, S. V., Kimchi-Sarfaty, C., Sauna, Z. E. & Gottesman, M. M. P-glycoprotein: from genomics to mechanism. Oncogene 22, 7468–7485 (2003).
Austin, R. P., Barton, P., Mohmed, S. & Riley, R. J. The binding of drugs to hepatocytes and its relationship to physicochemical properties. Drug Metab Dispos 33, 419–425 (2005).
Neuhoff, S., Ungell, A. L., Zamora, I. & Artursson, P. pH-dependent bidirectional transport of weakly basic drugs across Caco-2 monolayers: implications for drug-drug interactions. Pharm Res 20, 1141–1148 (2003).
Durazo, S. A., Kadam, R. S., Drechsel, D., Patel, M. & Kompella, U. B. Brain mitochondrial drug delivery: influence of drug physicochemical properties. Pharm Res 28, 2833–2847 (2011).
Hann, M. M. & Simpson, G. L. Intracellular drug concentration and disposition—the missing link? Methods 68, 283–285 (2014).
Dollery, C. T. Intracellular drug concentrations. Clin Pharmacol Ther 93, 263–266 (2013).
Gupta, A., Chatelain, P., Massingham, R., Jonsson, E. N. & Hammarlund-Udenaes, M. Brain distribution of cetirizine enantiomers: comparison of three different tissue-to-plasma partition coefficients: K(p), K(p,u), and K(p,uu). Drug Metab Dispos 34, 318–323 (2006).
Niemi, M., Pasanen, M. K., Neuvonen, P. J. Organic anion transporting polypeptide 1B1: a genetically polymorphic transporter of major importance for hepatic drug uptake. Pharmacol Rev 63, 157–181 (2011).
Metrabase: Metabolism and Transport Database (http://www-metrabase.ch.cam.ac.uk/).
DrugBank: Drug & Drug Target Database (http://www.drugbank.ca/).
Lipinski, C. A., Lombardo, F., Dominy, B. W. & Feeney, P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced Drug Delivery Reviews 23, 3–25 (1997).
Karlgren, M., Ahlin, G., Bergstrom, C. A., Svensson, R., Palm, J. & Artursson, P. In vitro and in silico strategies to identify OATP1B1 inhibitors and predict clinical drug-drug interactions. Pharm Res 29, 411–426 (2012).
Lecluyse, E. L. & Alexandre, E. Isolation and culture of primary hepatocytes from resected human liver tissue. Methods Mol Biol 640, 57–82 (2010).
Gillen, C. M. & Forbush, B., 3rd Functional interaction of the K-Cl cotransporter (KCC1) with the Na-K-Cl cotransporter in HEK-293 cells. Am J Physiol 276, C328–336 (1999).
Lu, C. et al. Comparison of intrinsic clearance in liver microsomes and hepatocytes from rats and humans: evaluation of free fraction and uptake in hepatocytes. Drug Metab Dispos 34, 1600–1605 (2006).
Nagaraj, N., Lu, A., Mann, M. & Wisniewski, J. R. Detergent-based but gel-free method allows identification of several hundred membrane proteins in single LC-MS runs. J Proteome Res 7, 5028–5032 (2008).
Groer, C. et al. Absolute protein quantification of clinically relevant cytochrome P450 enzymes and UDP-glucuronosyltransferases by mass spectrometry-based targeted proteomics. J Pharm Biomed Anal 100, 393–401 (2014).
Groer, C. et al. LC-MS/MS-based quantification of clinically relevant intestinal uptake and efflux transporter proteins. J Pharm Biomed Anal 85, 253–261 (2013).
Acknowledgements
The authors gratefully acknowledge SimulationsPlus for access to the ADMET Predictor software and ChemAxon for access to the JChem Suite. The authors also acknowledge the support from Science for Life Laboratory (SciLifeLab). We thank Stefan Oswald and Markus Keiser (University of Greifswald) for supplying the MDCK P-gp-transfected cell line. This work was supported by the Swedish Research Council (grants no. 2822 and 5715), the Swedish Fund for Research Without Animal Experiments, Carl Tryggers stiftelse, Magn. Bergvalls stiftelse, and Åke Wibergs stiftelse. André Mateus was supported by a PhD training grant from Fundação para a Ciência e Tecnologia (grant no. SFRH/BD/68304/2010). Andrea Treyer was supported by ARIADME, a European FP7 ITN Community’s Seventh Framework Program (grant no. 607517). Christine Wegler was supported by AstraZeneca.
Author information
Authors and Affiliations
Contributions
A.M. and A.T. performed all measurements of intracellular compound bioavailability. C.W. performed the proteomics measurements. M.K. established all cell lines used in this study. A.M., A.T., P.M. and P.A. performed the majority of data analysis and wrote the manuscript with critical input and contributions from all co-authors. A.M., A.T., P.M. and P.A. designed and supervised the study.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Mateus, A., Treyer, A., Wegler, C. et al. Intracellular drug bioavailability: a new predictor of system dependent drug disposition. Sci Rep 7, 43047 (2017). https://doi.org/10.1038/srep43047
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep43047
This article is cited by
-
Antidepressants enter cells, organelles, and membranes
Neuropsychopharmacology (2024)
-
A multiscale approach for bridging the gap between potency, efficacy, and safety of small molecules directed at membrane proteins
Scientific Reports (2021)
-
Lumbar cerebrospinal fluid-to-brain extracellular fluid surrogacy is context-specific: insights from LeiCNS-PK3.0 simulations
Journal of Pharmacokinetics and Pharmacodynamics (2021)
-
Enhancing intracellular accumulation and target engagement of PROTACs with reversible covalent chemistry
Nature Communications (2020)
-
Avocado-derived polyols for use as novel co-surfactants in low energy self-emulsifying microemulsions
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
